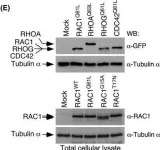
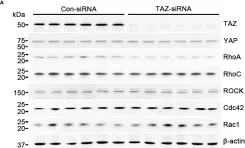
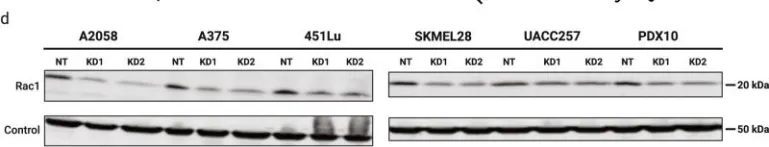
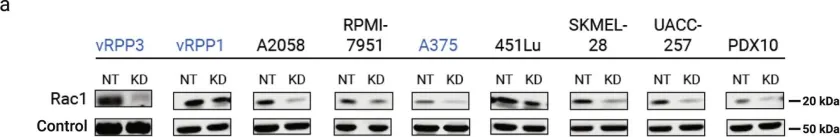


Podosomes are adhesion structures with densely-polymerized F-actin. While PI(3,4,5)P3 and Cdc42-GTP are known factors to trigger WASP-mediated actin polymerization at the macrophage podosome, their causal mechanism to activate WASP remains unclear. Here, we demonstrate that spatially elevated Cdc42-GTP is a downstream effector of local PI(3,4,5)P3 production at the podosome. We further examine the expression and distribution of 19 Cdc42 guanine exchange factors (GEFs) and identify VAV1 as the key PI(3,4,5)P3-dependent Cdc42 GEF. VAV1 is spatially enriched at the macrophage podosome, and the association of VAV1 with the membrane plays a critical role in upregulating its GEF activity. Reintroduction of wildtype VAV1, rather than the PI(3,4,5)P3-binding deficient or catalytically dead mutants restores the matrix degradation and chemotactic migration of VAV1-knockdown macrophage. Thus, the biogenesis of PI(3,4,5)P3 acts as an upstream signal to locally recruit VAV1 and in turn triggers the guanine nucleotide exchange of Cdc42. Elevated levels of Cdc42-GTP then promote WASP-mediated podosome assembly and macrophage chemotaxis.
© 2025. The Author(s).
Product Citations: 126
PI(3,4,5)P3-mediated Cdc42 activation regulates macrophage podosome assembly.
In Cellular and Molecular Life Sciences : CMLS on 24 March 2025 by Qi, Y. & Yu, C. H.
-
Biochemistry and Molecular biology
-
Immunology and Microbiology
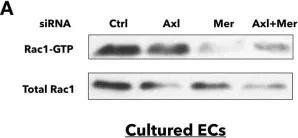
Plakoglobin does not participate in endothelial barrier stabilization mediated by cAMP.
In Scientific Reports on 16 March 2025 by Hamad, I., Šepić, S., et al.
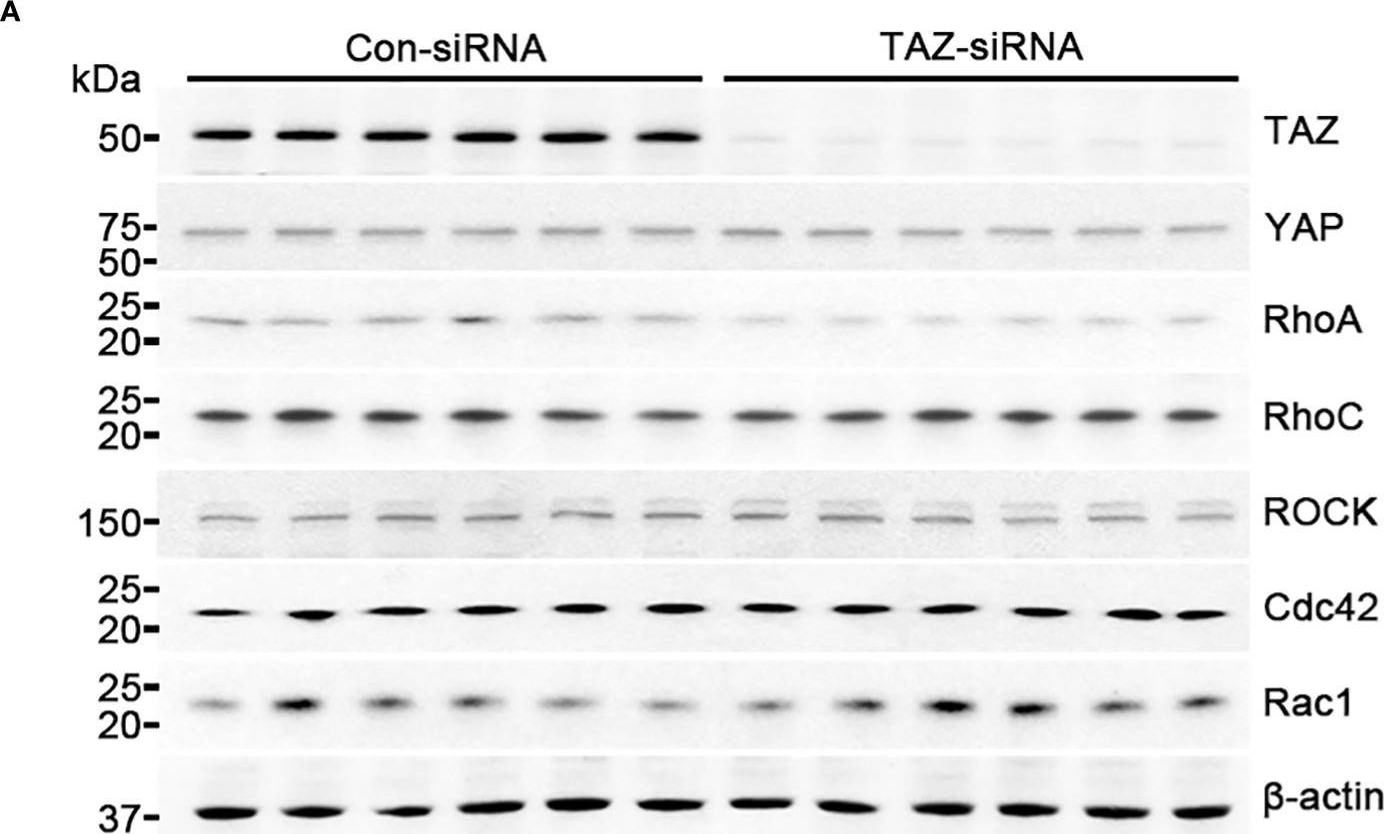
Critical for maintenance of endothelial barrier is the remodeling of the actin cytoskeleton and the precise control of junctional integrity. Plakoglobin (PG) is a structural and signaling protein involved in vascular permeability regulation together with key signaling molecules such as cAMP, Rho GTPases and actin-binding proteins. Here, we investigated the role of PG in cAMP-mediated endothelial barrier stabilization by establishing myocardial endothelial cells derived from wild type (WT) and PG knock-out (PG-KO) mice. Under basal conditions, TEER measurements showed increased barrier function of PG-KO, an effect associated with enhanced protein levels and junctional VE-cadherin and β-catenin accumulation. PG-KO cells also displayed more PECAM-1 and VE-PTP-phosphatase and less phosphorylated VE-cadherin, typically linked with modulation of junctional integrity. PG ablation neither changed the composition of VE-cadherin/β-catenin complex nor activities of Rac1 and RhoA but decreased the basal intracellular cAMP concentration. Remarkably, cAMP augmentation led to enhanced Rac1 activity and TEER in both cell lines, but the effect was less prominent in PG-KO. The tighter barrier in WT was paralleled with more VE-cadherin, β-catenin and cortactin, an actin-binding protein, towards junctions. Surprisingly, PG phosphorylation at Ser665 was not required for cAMP-mediated endothelial barrier integrity, which is different to cardiomyocyte and keratinocyte cell adhesion.
© 2025. The Author(s).
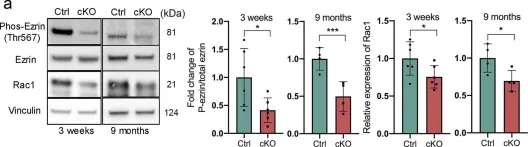
In Cells on 16 December 2024 by Lim, J., Hwang, Y. S., et al.
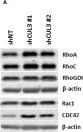
Rho guanine nucleotide dissociation inhibitor 1 (RhoGDI1) plays a critical role in regulating the activity of Rho guanosine triphosphatases (GTPases). Phosphorylation of RhoGDI1 dynamically modulates the activation of Rho GTPases, influencing cell proliferation and migration. This study explored the involvement of Never In Mitosis A (NIMA)-related serine/threonine protein kinase 2 (NEK2) in phosphorylating RhoGDI1 and its implications in cancer cell behavior associated with tumor progression. We employed GST pull-down assays and immunoprecipitation to investigate the interaction between NEK2 and RhoGDI1. Truncation fragments identified the region of RhoGDI1 responsible for binding with NEK2. Phosphorylation assays determined the site of NEK2-mediated phosphorylation on RhoGDI1. Functional assays were conducted using overexpression of the RhoGDI1 substitution mutant to assess their impact on cancer cell behavior. NEK2 directly bound to RhoGDI1 and phosphorylated it at Ser174. This phosphorylation event facilitated cancer cell proliferation and motility by activating RhoA and Rac1. The RhoGDI1 aa 112-134 region was critical for the binding to NEK2. Disruption of the NEK2-RhoGDI1 interaction through overexpression of a RhoGDI1 truncated fragment (aa 112-134) led to diminished RhoGDI1 phosphorylation and RhoA/Rac1 activation induced by NEK2, resulting in reduced cancer cell proliferation and migration. Moreover, in vivo studies showed reduced tumor growth and lung metastasis when the NEK2-RhoGDI1 interaction was disrupted. This study indicates that NEK2 promotes the metastatic behaviors of cancer cells by activating RhoA and Rac1 by phosphorylating RhoGDI1.
-
Cancer Research
-
Cell Biology
In Naunyn-Schmiedeberg's Archives of Pharmacology on 1 December 2024 by Matylitsky, J., Krieg, A., et al.
The dreaded nosocomial pathogen Clostridioides difficile causes diarrhea and severe inflammation of the colon, especially after the use of certain antibiotics. The bacterium releases two deleterious toxins, TcdA and TcdB, into the gut, which are mainly responsible for the symptoms of C. difficile-associated diseases (CDADs). Both toxins are capable of entering independently into various host cells, e.g., intestinal epithelial cells, where they mono-O-glucosylate and inactivate Rho and/or Ras GTPases, important molecular switches for various cellular functions. We have shown recently that the cellular uptake of the Clostridioides difficile toxins TcdA and TcdB (TcdA/B) is inhibited by the licensed class III antiarrhythmic drug amiodarone (Schumacher et al. in Gut Microbes 15(2):2256695, 2023). Mechanistically, amiodarone delays the cellular uptake of both toxins into target cells most likely by lowering membrane cholesterol levels and by interfering with membrane insertion and/or pore formation of TcdA/B. However, serious side effects, such as thyroid dysfunction and severe pulmonary fibrosis, limit the clinical use of amiodarone in patients with C. difficile infection (CDI). For that reason, we aimed to test whether dronedarone, an amiodarone derivative with a more favorable side effect profile, is also capable of inhibiting TcdA/B. To this end, we tested in vitro with various methods the impact of dronedarone on the intoxication of Vero and CaCo-2 cells with TcdA/B. Importantly, preincubation of both cell lines with dronedarone for 1 h at concentrations in the low micromolar range rendered the cells less sensitive toward TcdA/B-induced Rac1 glucosylation, collapse of the actin cytoskeleton, cell rounding, and cytopathic effects, respectively. Our study points toward the possibility of repurposing the already approved drug dronedarone as the preferable safer-to-use alternative to amiodarone for inhibiting TcdA/B in the (supportive) therapy of CDADs.
© 2024. The Author(s).
-
WB
-
Pharmacology
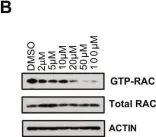
In Molecules (Basel, Switzerland) on 9 November 2024 by Carmona-Carmona, C. A., Zini, P., et al.
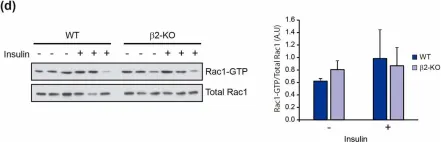
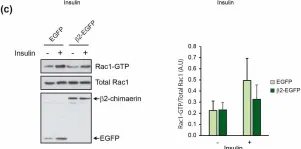
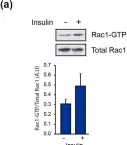
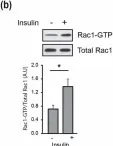
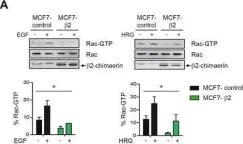
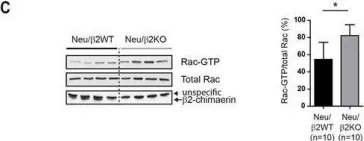
Glucose homeostasis is a complex process regulated by multiple organs and hormones, with insulin playing a central role. Recent evidence underscores the role of small GTP-binding proteins, particularly Rac1, in regulating insulin secretion and glucose uptake. However, the role of Rac1-regulatory proteins in these processes remains largely unexplored. In this study, we investigated the role of β2-chimaerin, a Rac1-specific GTPase-activating protein (GAP), in glucose homeostasis using whole-body β2-chimaerin knockout mice. Our data revealed that β2-chimaerin deficiency results in improved glucose tolerance and enhanced insulin sensitivity in mice. These metabolic effects were associated with increased insulin-induced AKT phosphorylation in the liver and activation of downstream pathways that regulate gluconeogenesis and glycogen synthesis. We show that insulin activates Rac1 in the liver. However, β2-chimaerin deletion did not significantly alter Rac1 activation in this organ, suggesting that β2-chimaerin regulates insulin signaling via a Rac1-independent mechanism. These findings expand our understanding of Rac1 regulation in glucose metabolism, and identify β2-chimaerin as a novel modulator of hepatic insulin signaling, with potential implications for the development of insulin resistance and diabetes.
-
WB
-
Biochemistry and Molecular biology
-
Cell Biology
-
Endocrinology and Physiology
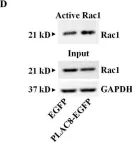
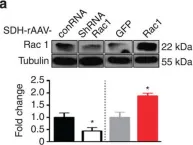
In Molecules on 9 November 2024 by Carmona-Carmona, C. A., Zini, P., et al.
Fig.5.D

-
WB
-
Collected and cropped from Molecules by CiteAb, provided under a CC-BY license
Image 1 of 31
In Molecules on 9 November 2024 by Carmona-Carmona, C. A., Zini, P., et al.
Fig.5.C

-
WB
-
Collected and cropped from Molecules by CiteAb, provided under a CC-BY license
Image 1 of 31
In Molecules on 9 November 2024 by Carmona-Carmona, C. A., Zini, P., et al.
Fig.5.A

-
WB
-
Collected and cropped from Molecules by CiteAb, provided under a CC-BY license
Image 1 of 31
In Molecules on 9 November 2024 by Carmona-Carmona, C. A., Zini, P., et al.
Fig.5.B

-
WB
-
Collected and cropped from Molecules by CiteAb, provided under a CC-BY license
Image 1 of 31
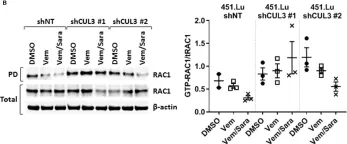
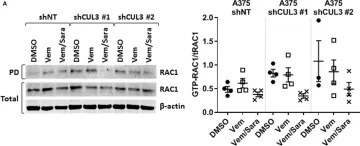
In Mol Oncol on 9 August 2024 by Conde, J., Fernández-Pisonero, I., et al.
Fig.2.E

-
WB
-
Collected and cropped from Mol Oncol by CiteAb, provided under a CC-BY license
Image 1 of 31
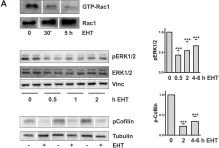
In Front Oncol on 22 May 2024 by Choi, H. S., Jang, H. J., et al.
Fig.4.A
-
WB
-
Homo sapiens (Human)
Collected and cropped from Front Oncol by CiteAb, provided under a CC-BY license
Image 1 of 31
In NPJ Precis Oncol on 21 October 2022 by Zhu, E. Y., Riordan, J. D., et al.
Fig.1.D

-
WB
-
Collected and cropped from NPJ Precis Oncol by CiteAb, provided under a CC-BY license
Image 1 of 31
In NPJ Precis Oncol on 21 October 2022 by Zhu, E. Y., Riordan, J. D., et al.
Fig.1.A

-
WB
-
Collected and cropped from NPJ Precis Oncol by CiteAb, provided under a CC-BY license
Image 1 of 31
In NPJ Precis Oncol on 21 October 2022 by Zhu, E. Y., Riordan, J. D., et al.
Fig.3.C

-
WB
-
Collected and cropped from NPJ Precis Oncol by CiteAb, provided under a CC-BY license
Image 1 of 31
In NPJ Precis Oncol on 21 October 2022 by Zhu, E. Y., Riordan, J. D., et al.
Fig.3.D

-
WB
-
Collected and cropped from NPJ Precis Oncol by CiteAb, provided under a CC-BY license
Image 1 of 31
In Blood Cancer J on 14 April 2022 by Canovas Nunes, S., De Vita, S., et al.
Fig.2.B

-
WB
-
Collected and cropped from Blood Cancer J by CiteAb, provided under a CC-BY license
Image 1 of 31
In Cancers (Basel) on 5 January 2022 by Kim, H. J., Ryu, K. J., et al.
Fig.3.A

-
WB
-
Collected and cropped from Cancers (Basel) by CiteAb, provided under a CC-BY license
Image 1 of 31
In Commun Biol on 8 July 2021 by Shang, P., Stepicheva, N., et al.
Fig.2.A

-
WB
-
Collected and cropped from Commun Biol by CiteAb, provided under a CC-BY license
Image 1 of 31
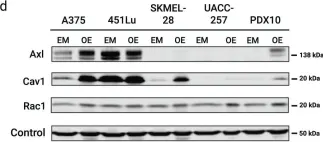
In Front Oncol on 30 April 2020 by Vanneste, M., Feddersen, C. R., et al.
Fig.5.B

-
WB
-
Homo sapiens (Human)
Collected and cropped from Front Oncol by CiteAb, provided under a CC-BY license
Image 1 of 31
In Front Oncol on 30 April 2020 by Vanneste, M., Feddersen, C. R., et al.
Fig.6.A

-
WB
-
Homo sapiens (Human)
Collected and cropped from Front Oncol by CiteAb, provided under a CC-BY license
Image 1 of 31
In Front Oncol on 30 April 2020 by Vanneste, M., Feddersen, C. R., et al.
Fig.5.A

-
WB
-
Homo sapiens (Human)
Collected and cropped from Front Oncol by CiteAb, provided under a CC-BY license
Image 1 of 31
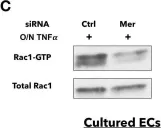
In PLoS One on 6 December 2019 by Li, Y., Wittchen, E. S., et al.
Fig.6.A

-
WB
-
Collected and cropped from PLoS One by CiteAb, provided under a CC-BY license
Image 1 of 31
In PLoS One on 6 December 2019 by Li, Y., Wittchen, E. S., et al.
Fig.6.C

-
WB
-
Collected and cropped from PLoS One by CiteAb, provided under a CC-BY license
Image 1 of 31
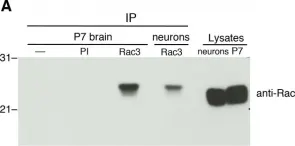
In PLoS One on 2 August 2019 by Pennucci, R., Gucciardi, I., et al.
Fig.1.A

-
WB
-
Mus musculus (House mouse)
Collected and cropped from PLoS One by CiteAb, provided under a CC-BY license
Image 1 of 31
In Development on 29 January 2018 by Chang, W. L., Liu, Y. W., et al.
Fig.6.D

-
WB
-
Collected and cropped from Development by CiteAb, provided under a CC-BY license
Image 1 of 31
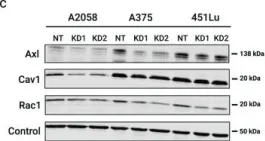
In Oncotarget on 10 May 2016 by Casado-Medrano, V., Barrio-Real, L., et al.
Fig.1.A

-
WB
-
Collected and cropped from Oncotarget by CiteAb, provided under a CC-BY license
Image 1 of 31
In Oncotarget on 10 May 2016 by Casado-Medrano, V., Barrio-Real, L., et al.
Fig.7.C

-
WB
-
Collected and cropped from Oncotarget by CiteAb, provided under a CC-BY license
Image 1 of 31
In Nat Commun on 19 April 2016 by Blas-Rus, N., Bustos-Morán, E., et al.
Fig.5.B

-
WB
-
Homo sapiens (Human)
Collected and cropped from Nat Commun by CiteAb, provided under a CC-BY license
Image 1 of 31
In Nat Commun on 13 April 2015 by Lu, J., Luo, C., et al.
Fig.4.A

-
WB
-
Collected and cropped from Nat Commun by CiteAb, provided under a CC-BY license
Image 1 of 31
In PLoS One on 31 March 2015 by Giehl, K., Keller, C., et al.
Fig.7.A

-
WB
-
Collected and cropped from PLoS One by CiteAb, provided under a CC-BY license
Image 1 of 31