Procaspase 9 is the initiator caspase for apoptosis, but how its levels and activities are maintained remains unclear. The gigantic Inhibitor-of-Apoptosis Protein BIRC6/BRUCE/Apollon inhibits both apoptosis and autophagy by promoting ubiquitylation of proapoptotic factors and the key autophagic protein LC3, respectively. Here we show that BIRC6 forms an anti-parallel U-shaped dimer with multiple previously unannotated domains, including a ubiquitin-like domain, and the proapoptotic factor Smac/DIABLO binds BIRC6 in the central cavity. Notably, Smac outcompetes the effector caspase 3 and the pro-apoptotic protease HtrA2, but not procaspase 9, for binding BIRC6 in cells. BIRC6 also binds LC3 through its LC3-interacting region, probably following dimer disruption of this BIRC6 region. Mutation at LC3 ubiquitylation site promotes autophagy and autophagic degradation of BIRC6. Moreover, induction of autophagy promotes autophagic degradation of BIRC6 and caspase 9, but not of other effector caspases. These results are important to understand how the balance between apoptosis and autophagy is regulated under pathophysiological conditions.
© 2024. The Author(s).
Product Citations: 32
Molecular mechanisms underlying the BIRC6-mediated regulation of apoptosis and autophagy.
In Nature Communications on 30 January 2024 by Liu, S. S., Jiang, T. X., et al.
-
WB
-
Cell Biology
ARTS and small-molecule ARTS mimetics upregulate p53 levels by promoting the degradation of XIAP
Preprint on Research Square on 1 December 2023 by Abbas, R., Hartmann, O., et al.
Mutations resulting in decreased activity of p53 tumor suppressor protein promote tumorigenesis. P53 protein levels are tightly regulated through the Ubiquitin Proteasome System (UPS). Several E3 ligases were shown to regulate p53 stability, including MDM2. Here we report that the ubiquitin E3 ligase XIAP (X-linked Inhibitors of Apoptosis) is a direct ligase for p53 and describe a novel approach for modulating the levels of p53 by targeting the XIAP pathway. Using in vivo (live-cell) and in vitro (cell-free reconstituted system) ubiquitylation assays, we show that the XIAP-antagonist ARTS regulates the levels of p53 by promoting the degradation of XIAP. XIAP directly binds and ubiquitylates p53. In apoptotic cells, ARTS inhibits the ubiquitylation of p53 by antagonizing XIAP. XIAP knockout MEFs express higher p53 protein levels compared to wild-type MEFs. Computational screen for small molecules with high affinity to the ARTS-binding site within XIAP identified a small-molecule ARTS-mimetic, B3. This compound stimulates apoptosis in a wide range of cancer cells but not normal PBMC (Peripheral Blood Mononuclear Cells ) . Like ARTS, the B3 compound binds to XIAP and promotes its degradation via the UPS. B3 binding to XIAP stabilizes p53 by disrupting its interaction with XIAP. These results reveal a novel mechanism by which ARTS and p53 regulate each other through an amplification loop to promote apoptosis. Finally, these data suggest that targeting the ARTS binding pocket in XIAP can be used to increase p53 levels as a new strategy for developing anti-cancer therapeutics.
Constitutive protein degradation induces acute cell death via proteolysis products
Preprint on BioRxiv : the Preprint Server for Biology on 6 February 2023 by Chen, S., Prakash, S., et al.
Modulation of proteolysis is an emerging therapeutic mainstay. The clinical success of thalidomide and analogs has inspired development of rationally-designed therapeutics that repurpose endogenous degradation machinery to target pathogenic proteins. However, it is unknown whether target removal is the critical effect that drives degrader-induced efficacy. Here we report that proteasome-generated peptides actively initiate degrader-induced cell death. Utilizing BET family degraders as exemplars, we find that induced proteasomal degradation of the BRD4-long isoform (BRD4-L) generates neo-amino-terminal peptides that neutralize Inhibitor of Apoptosis (IAP) proteins to precipitate cell death. Depletion of BRD4-L paradoxically suppresses caspase activation induced by numerous BET degraders. An unbiased screen revealed that other degrader compounds, including clinical CELMoDs, rely on the same mechanism to potentiate caspase activation and apoptosis. Finally, in the context of constitutive immunoglobulin proteostasis within multiple myeloma cells, we report that therapeutic proteasomal protease inhibition alters the peptide repertoire to neutralize IAPs, thus contributing to the clinical efficacy of bortezomib. Together, these findings clarify the counterintuitive clinical benefit achieved by combining thalidomide analogs with proteasome inhibitors. Our study reveals a previously unrealized pro-apoptotic function of the peptides generated by a variety of proteolysis-modulating compounds, that provide design considerations to maximize therapeutic benefit.
Structural basis for BIRC6 to balance apoptosis and autophagy
Preprint on BioRxiv : the Preprint Server for Biology on 11 December 2022 by Liu, S., Jiang, T., et al.
ABSTRACT Caspase-9 is the initiator caspase for the intrinsic apoptotic cell death pathway, and is critical to the activation of effector caspases during apoptosis, but how its levels and activities are maintained remains unclear. The gigantic Inhibitor of Apoptosis Protein (IAP) BIRC6/BRUCE/Apollon not only inhibits apoptosis, but also promotes ubiquitination of the key autophagic protein LC3 and inhibits autophagy. Here we show that BIRC6 forms an anti-parallel U-shaped dimer in a 3.6-Å cryo-EM structure with multiple previously unannotated domains, including a ubiquitin-like domain, and discover that the mitochondria-derived pro-apoptotic factor Smac/DIABLO binds BIRC6 by interacting with one BIR domain, two carbohydrate-binding modules and two helices in the central cavity. Notably, Smac outcompetes the effector caspase 3 and the pro-apoptotic protease HtrA2, but not caspase 9, for binding BIRC6. BIRC6 strongly inhibits cellular activity of caspase 9, but weakly suppresses that of caspase 3. Meanwhile, BIRC6 binds LC3 through an LC3-interacting region, probably following dimer disruption of this BIRC6 region. Deficiency in LC3 ubiquitination promotes autophagy and autophagic degradation of BIRC6, and inhibits apoptosis. Moreover, induction of autophagy promotes autophagic degradation of both procaspase-9 and active caspase-9, but not of effector caspases. These results are important to understand how the balance between apoptosis and autophagy is regulated under pathophysiological conditions.
-
WB
-
Cell Biology
In Oncology Letters on 1 November 2022 by Jang, J. H., Lee, T. J., et al.
Dapagliflozin is a sodium/glucose cotransporter 2 inhibitor used recently to treat patients with type 2 diabetes. A recent study has demonstrated that dapagliflozin induces apoptosis in human renal and breast tumor cells. However, to the best of our knowledge, the molecular mechanism underlying dapagliflozin-mediated apoptosis in Caki-1 human renal carcinoma cells has not been elucidated. The present study demonstrated that the dapagliflozin treatment dose-dependently increased cell death in Caki-1 cells. Dapagliflozin treatment also induced apoptosis as confirmed by FITC-conjugated Annexin V/PI staining. Additionally, treatment with dapagliflozin reduced the expression levels of anti-apoptotic proteins, cellular Fas-associated death domain-like interleukin-1-converting enzyme-inhibitory protein (cFLIP)L and cFLIPS in Caki-1 cells. Benzyloxycarbonyl-Val-Ala-Asp-fluoromethyl-ketone inhibited dapagliflozin-induced apoptosis, implying that dapagliflozin-induced apoptosis is regulated by a caspase-dependent pathway. By contrast, N-acetylcysteine had no effect on dapagliflozin-induced apoptosis and downregulation of cFLIPL and cFLIPS expression. Furthermore, overexpression of cFLIPL, but not cFLIPS, partially inhibited apoptosis induced by dapagliflozin. cFLIPL and cFLIPS mRNA levels remained constant in Caki-1 cells after treatment with 0, 20, 40, 60, 80 and 100 µM dapagliflozin. Notably, it was confirmed that cFLIPS protein levels were reduced due to the increased cFLIPS instability in dapagliflozin-treated Caki-1 cells. The present study also demonstrated that dapagliflozin had no effect on HK-2 normal human kidney cells. Taken together, the present study revealed that dapagliflozin induced apoptosis via the downregulation of cFLIPL and an increase in cFLIPS instability, suggesting that dapagliflozin may be a feasible drug candidate for the treatment of human renal cancer.
Copyright: © Jang et al.
-
WB
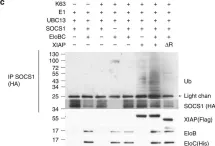
In Commun Biol on 24 April 2020 by Wu, X., Gardashova, G., et al.
Fig.1.C

-
WB
-
Collected and cropped from Commun Biol by CiteAb, provided under a CC-BY license
Image 1 of 7
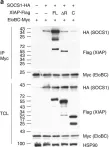
In Commun Biol on 24 April 2020 by Wu, X., Gardashova, G., et al.
Fig.8.B

-
WB
-
Collected and cropped from Commun Biol by CiteAb, provided under a CC-BY license
Image 1 of 7
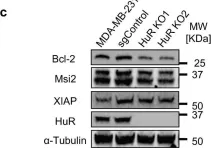
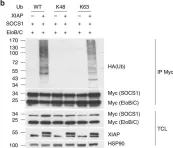
In Nat Commun on 31 January 2018 by Hsieh, W. C., Hsu, T. S., et al.
Fig.4.C

-
WB
-
Homo sapiens (Human)
Collected and cropped from Nat Commun by CiteAb, provided under a CC-BY license
Image 1 of 7
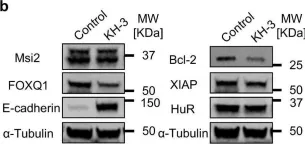
In Nat Commun on 31 January 2018 by Hsieh, W. C., Hsu, T. S., et al.
Fig.4.A

-
WB
-
Homo sapiens (Human)
Collected and cropped from Nat Commun by CiteAb, provided under a CC-BY license
Image 1 of 7
In Nat Commun on 31 January 2018 by Hsieh, W. C., Hsu, T. S., et al.
Fig.4.B

-
WB
-
Homo sapiens (Human)
Collected and cropped from Nat Commun by CiteAb, provided under a CC-BY license
Image 1 of 7
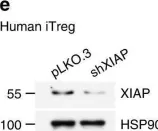
In Nat Commun on 31 January 2018 by Hsieh, W. C., Hsu, T. S., et al.
Fig.1.E

-
WB
-
Homo sapiens (Human)
Collected and cropped from Nat Commun by CiteAb, provided under a CC-BY license
Image 1 of 7
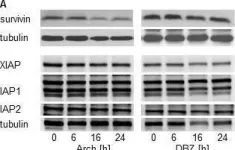
In Oncotarget on 22 December 2015 by Zhang, S., Schneider, L. S., et al.
Fig.8.A

-
WB
-
Collected and cropped from Oncotarget by CiteAb, provided under a CC-BY license
Image 1 of 7