
Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) is a cytotoxic cytokine that induces cancer cell death by binding to TRAIL receptors. Because of its selective cytotoxicity toward cancer cells, TRAIL therapeutics, such as recombinant TRAIL and agonistic antibodies targeting TRAIL receptors, have garnered attention as promising cancer treatment agents. However, many cancer cells acquire resistance to TRAIL-induced cell death. To overcome this issue, we searched for agents to sensitize cancer cells to TRAIL-induced cell death by screening a small-molecule chemical library consisting of diverse compounds. We identified a cardiac glycoside, proscillaridin A, as the most effective TRAIL sensitizer in colon cancer cells. Proscillaridin A synergistically enhanced TRAIL-induced cell death in TRAIL-sensitive and -resistant colon cancer cells. Additionally, proscillaridin A enhanced cell death in cells treated with TRAIL and TRAIL sensitizer, the second mitochondria-derived activator of caspase mimetic. Proscillaridin A upregulated TRAIL receptor expression, while downregulating the levels of the anti-cell death molecules, cellular FADD-like IL-1β converting enzyme-like inhibitor protein and Mcl1, in a cell type-dependent manner. Furthermore, proscillaridin A enhanced TRAIL-induced cell death partly via O-glycosylation. Taken together, our findings suggest that proscillaridin A is a promising agent that enhances the anti-cancer efficacy of TRAIL therapeutics.
Product Citations: 12
Proscillaridin A Sensitizes Human Colon Cancer Cells to TRAIL-Induced Cell Death.
In International Journal of Molecular Sciences on 23 June 2022 by Semba, M., Takamatsu, S., et al.
-
WB
-
Homo sapiens (Human)
-
Cancer Research
In Cancer Cell on 13 May 2019 by Elgendy, M., Cirò, M., et al.
Tumor cells may adapt to metabolic challenges by alternating between glycolysis and oxidative phosphorylation (OXPHOS). To target this metabolic plasticity, we combined intermittent fasting, a clinically feasible approach to reduce glucose availability, with the OXPHOS inhibitor metformin. In mice exposed to 24-h feeding/fasting cycles, metformin impaired tumor growth only when administered during fasting-induced hypoglycemia. Synergistic anti-neoplastic effects of the metformin/hypoglycemia combination were mediated by glycogen synthase kinase 3β (GSK3β) activation downstream of PP2A, leading to a decline in the pro-survival protein MCL-1, and cell death. Mechanistically, specific activation of the PP2A-GSK3β axis was the sum of metformin-induced inhibition of CIP2A, a PP2A suppressor, and of upregulation of the PP2A regulatory subunit B56δ by low glucose, leading to an active PP2A-B56δ complex with high affinity toward GSK3β.
Copyright © 2019 Elsevier Inc. All rights reserved.
-
Biochemistry and Molecular biology
-
Cancer Research
-
Cell Biology
In Clinical Cancer Research on 1 March 2019 by Andrews, A., Warner, K., et al.
Unique cells characterized by multipotency, self-renewal, and high tumorigenic potential have been recently discovered in mucoepidermoid carcinomas. These cells are defined by high aldehyde dehydrogenase activity and high CD44 expression (ALDHhighCD44high) and function as cancer stem cells (CSC). It has been recently shown that p53 regulates cell differentiation, suggesting that induction of p53 by therapeutic blockade of the MDM2-p53 interaction may constitute a novel strategy to ablate CSCs. Here, we evaluated the effect of a small-molecule inhibitor of MDM2-p53 interaction (MI-773) on the fraction of CSCs in mucoepidermoid carcinoma.
Human mucoepidermoid carcinoma cells (UM-HMC-1,-3A,-3B) were used to assess the effect of MI-773 on cell survival, cell cycle, fraction of CSCs, and expression of p53, p21, MDM2, and Bmi-1 (key regulator of self-renewal). Mice bearing xenograft tumors generated with these mucoepidermoid carcinoma cells were treated with MI-773 to determine the effect of MDM2-p53 inhibition on CSCs in vivo.
MDM2 is highly expressed in human mucoepidermoid carcinoma tissues. MI-773 induced expression of p53 and its downstream targets p21 and MDM2, caused G1 cell-cycle arrest, and induced mucoepidermoid carcinoma tumor cell apoptosis in vitro. Importantly, a marked decrease in expression of Bmi-1 and in the fraction of ALDHhighCD44high (CSCs) was caused by MI-773 in vitro and in mice harboring mucoepidermoid carcinoma xenografts.
Collectively, these data demonstrate that MI-773 reduces the fraction of CSCs, suggesting that patients with mucoepidermoid carcinoma might benefit from therapeutic inhibition of the MDM2-p53 interaction.
©2018 American Association for Cancer Research.
-
WB
-
Cancer Research
-
Stem Cells and Developmental Biology
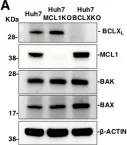
Infection with flaviviruses requires BCLXL for cell survival.
In PLoS Pathogens on 1 September 2018 by Suzuki, T., Okamoto, T., et al.
BCL2 family proteins including pro-survival proteins, BH3-only proteins and BAX/BAK proteins control mitochondria-mediated apoptosis to maintain cell homeostasis via the removal of damaged cells and pathogen-infected cells. In this study, we examined the roles of BCL2 proteins in the induction of apoptosis in cells upon infection with flaviviruses, such as Japanese encephalitis virus, Dengue virus and Zika virus. We showed that survival of the infected cells depends on BCLXL, a pro-survival BCL2 protein due to suppression of the expression of another pro-survival protein, MCL1. Treatment with BCLXL inhibitors, as well as deficient BCLXL gene expression, induced BAX/BAK-dependent apoptosis upon infection with flaviviruses. Flavivirus infection attenuates cellular protein synthesis, which confers reduction of short-half-life proteins like MCL1. Inhibition of BCLXL increased phagocytosis of virus-infected cells by macrophages, thereby suppressing viral dissemination and chemokine production. Furthermore, we examined the roles of BCLXL in the death of JEV-infected cells during in vivo infection. Haploinsufficiency of the BCLXL gene, as well as administration of BH3 mimetic compounds, increased survival rate after challenge of JEV infection and suppressed inflammation. These results suggest that BCLXL plays a crucial role in the survival of cells infected with flaviviruses, and that BCLXL may provide a novel antiviral target to suppress propagation of the family of Flaviviridae viruses.
-
WB
-
Homo sapiens (Human)
-
Immunology and Microbiology
In BMC Cancer on 6 February 2018 by Lu, C., Yang, D., et al.
Pancreas ductal adenocarcinoma (PDAC) has the most dismal prognosis among all human cancers since it is highly resistant to chemotherapy, radiotherapy and immunotherapy. The anticipated consequence of all therapies is induction of tumor apoptosis. The highly resistance nature of PDACs to all therapies suggests that the intrinsic tumor cell factors, likely the deregulated apoptosis pathway, are key mechanisms underlying PDAC non-response to these therapies, rather than the therapeutic agents themselves. The aim of this study is to test the hypothesis that epigenetic dysregulation of apoptosis mediators underlies PDAC resistance to gemcitabine, the standard chemotherapy for human PDAC.
PDAC cells were analyzed for apoptosis sensitivity in the presence of a selective epigenetic inhibitor. The epigenetic regulation of apoptosis regulators was determined by Western Blotting and quantitative PCR. The specific epigenetic modification of apoptosis regulator promoter chromatin was determined by chromatin immunoprecipitation in PDAC cells.
Inhibition of histone methyltransferase (HMTase) by a selective HMTase inhibitor, verticillin A, significantly increased human PDAC cell sensitivity to gemcitabine-induced growth suppression. Verticillin A treatment decreased FLIP, Mcl-1, Bcl-x and increased Bak, Bax and Bim protein level in the tumor cells, resulting in activation of caspases, elevated cytochrome C release and increased apoptosis as determined by upregulated PARP cleavage in tumor cells. Analysis of human PDAC specimens indicated that the expression levels of anti-apoptotic mediators Bcl-x, Mcl-1, and FLIP were significantly higher, whereas the expression levels of pro-apoptotic mediators Bim, Bak and Bax were dramatically lower in human PDAC tissues as compared to normal pancreas. Verticillin A downregulated H3K4me3 levels at the BCL2L1, CFLAR and MCL-1 promoter to decrease Bcl-x, FLIP and Mcl-1 expression level, and inhibited H3K9me3 levels at the BAK1, BAX and BCL2L11 promoter to upregulate Bak, Bax and Bim expression level.
We determined that PDAC cells use H3K4me3 to activate Bcl-x, FLIP and Mcl-1, and H3K9me3 to silence Bak, Bax and Bim to acquire an apoptosis-resistant phenotype. Therefore, selective inhibition of H3K4me3 and H3K9me3 is potentially an effective approach to overcome PDAC cells resistance to gemcitabine.
-
WB
-
Homo sapiens (Human)
-
Cancer Research
In Int J Mol Sci on 23 June 2022 by Semba, M., Takamatsu, S., et al.
Fig.4.C

-
WB
-
Homo sapiens (Human)
Collected and cropped from Int J Mol Sci by CiteAb, provided under a CC-BY license
Image 1 of 2
In PLoS Pathog on 1 September 2018 by Suzuki, T., Okamoto, T., et al.
Fig.2.A

-
WB
-
Homo sapiens (Human)
Collected and cropped from PLoS Pathog by CiteAb, provided under a CC-BY license
Image 1 of 2