Chaperone-mediated autophagy (CMA) is a chaperone-dependent process of selective cytosolic protein turnover that targets specific proteins to lysosomes for degradation. Enhancing protein degradation mechanisms has been shown to be beneficial in multiple models of cardiac disease, including myocardial infarction (MI) and ischemia-reperfusion (I/R) injury. However, the causal role of CMA in cardiomyocyte injury and death is largely unknown. Hypoxia is an important contributor to both MI and I/R damage, which are major, precedent causes of heart failure. Upregulating CMA was hypothesized to protect against hypoxia-induced cardiomyocyte death. Lysosome-associated membrane protein 2a (Lamp2a) overexpression and knockdown were used to causally study CMA's role in hypoxically stressed cardiomyocytes. LAMP2a protein levels were used as both a primary indicator and driver of CMA function. Hypoxic stress was stimulated by CoCl2 treatment, which increased LAMP2a protein levels (+1.4-fold) and induced cardiomyocyte apoptosis (+3.2-4.0-fold). Lamp2a siRNA knockdown (-3.2-fold) of control cardiomyocytes increased apoptosis (+1.8-fold) suggesting that loss of CMA is detrimental for cardiomyocyte survival. However, there was neither an additive nor a synergistic effect on cell death when Lamp2a-silenced cells were treated with CoCl2. Conversely, Lamp2a overexpression (+3.0-fold) successfully reduced hypoxia-induced apoptosis by ∼50%. LAMP2a was also significantly increased (+1.7-fold) in ischemic heart failure patient samples, similar to hypoxically stressed cardiomyocytes. The failing ischemic hearts may have had insufficient CMA activation. To our knowledge, this study for the first time establishes a protective role for CMA (via Lamp2a overexpression) against hypoxia-induced cardiomyocyte loss and reveals the intriguing possibility that CMA activation may offer a cardioprotective treatment for ischemic heart disease.
Product Citations: 10
Chaperone-mediated autophagy protects cardiomyocytes against hypoxic-cell death.
In American Journal of Physiology - Cell Physiology on 1 November 2022 by Ghosh, R., Gillaspie, J. J., et al.
-
Cell Biology
-
Endocrinology and Physiology
In International Journal of Clinical and Experimental Pathology on 15 June 2022 by Xu, Q., Gao, B., et al.
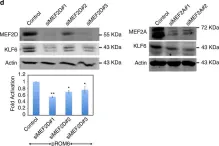
Hepatocellular carcinoma (HCC) is one of the deadliest malignancies in the world. There is a lack of effective treatment. Previous studies have shown that myocyte enhancer factor 2D (MEF2D) promotes the progression of HCC. Underlying mechanisms have not been fully elucidated. In this study, we reported experimental results obtained using double luciferase. Our results showed that AMOTL2, a negative regulator of Hippo/YAP signaling, and the MEF2 cis-acting element in the upstream region of its promoter bind to MEF2D, inhibiting its transcriptional expression. Studies confirmed that MEF2D affected the protein expression level of AMOTL2 and the YAP signaling activation. It promoted the migration and proliferation of hepatoma cells. We found that luteolin, a natural flavonoid, has anti-tumor activity in HCC cells by affecting YAP signaling transduction. In conclusion, we demonstrated that AMOTL2/YAP signaling is associated with MEF2D-related HCC progression. Luteolin is a promising anti-HCC compound for regulating this signaling.
IJCEP Copyright © 2022.
-
WB
-
Homo sapiens (Human)
-
Cancer Research
-
Pathology
In The Journal of Clinical Investigation on 1 June 2022 by Lu, Z., Mao, W., et al.
Poly(ADP-ribose) polymerase inhibitors (PARP inhibitors) have had an increasing role in the treatment of ovarian and breast cancers. PARP inhibitors are selectively active in cells with homologous recombination DNA repair deficiency caused by mutations in BRCA1/2 and other DNA repair pathway genes. Cancers with homologous recombination DNA repair proficiency respond poorly to PARP inhibitors. Cancers that initially respond to PARP inhibitors eventually develop drug resistance. We have identified salt-inducible kinase 2 (SIK2) inhibitors, ARN3236 and ARN3261, which decreased DNA double-strand break (DSB) repair functions and produced synthetic lethality with multiple PARP inhibitors in both homologous recombination DNA repair deficiency and proficiency cancer cells. SIK2 is required for centrosome splitting and PI3K activation and regulates cancer cell proliferation, metastasis, and sensitivity to chemotherapy. Here, we showed that SIK2 inhibitors sensitized ovarian and triple-negative breast cancer (TNBC) cells and xenografts to PARP inhibitors. SIK2 inhibitors decreased PARP enzyme activity and phosphorylation of class-IIa histone deacetylases (HDAC4/5/7). Furthermore, SIK2 inhibitors abolished class-IIa HDAC4/5/7-associated transcriptional activity of myocyte enhancer factor-2D (MEF2D), decreasing MEF2D binding to regulatory regions with high chromatin accessibility in FANCD2, EXO1, and XRCC4 genes, resulting in repression of their functions in the DNA DSB repair pathway. The combination of PARP inhibitors and SIK2 inhibitors provides a therapeutic strategy to enhance PARP inhibitor sensitivity for ovarian cancer and TNBC.
-
WB
In Physiological Genomics on 1 January 2018 by Li, L., Rubin, L. P., et al.
Development of the human placenta and its trophoblast cell types is critical for a successful pregnancy. Defects in trophoblast invasion and differentiation are associated with adverse pregnancy outcomes, including preeclampsia. The members of myocyte enhancer factor-2 (MEF2) family of transcription factors are key regulators of cellular proliferation, differentiation, and invasion in various cell types and tissues and might play a similarly important role in regulating trophoblast proliferation, invasion, and differentiation during human placental development. In the present study, using human cytotrophoblast cell lines (HTR8/SVneo and BeWo) and primary human cytotrophoblasts (CTBs), we show that members of the MEF2 family are differentially expressed in human placental CTBs, with MEF2B and MEF2D being highly expressed in first trimester extravillous CTBs. Overexpression of MEF2D results in cytotrophoblast proliferation and enhances the invasion and migration of extravillous-like HTR8/SVneo cells. This invasive property is blocked by overexpression of a dominant negative MEF2 (dnMEF2). In contrast, MEF2A is the principal MEF2 isoform expressed in term CTBs, MEF2C and MEF2D being expressed more weakly, and MEF2B expression being undetected. Overexpression of MEF2A induces cytotrophoblast differentiation and syncytium formation in BeWo cells. During in vitro differentiation of primary CTBs, MEF2A expression is associated with CTB differentiation into syncytiotrophoblast. Additionally, the course of p38 MAPK and ERK5 activities parallels the increase in MEF2A expression. These findings suggest individual members of MEF2 family distinctively regulate cytotrophoblast proliferation, invasion, and differentiation. Dysregulation of expression of MEF2 family or of their upstream signaling pathways may be associated with placenta-related pregnancy disorders.
-
WB
-
Homo sapiens (Human)
-
Biochemistry and Molecular biology
Phosphorylation of LAMP2A by p38 MAPK couples ER stress to chaperone-mediated autophagy.
In Nature Communications on 24 November 2017 by Li, W., Zhu, J., et al.
Endoplasmic reticulum (ER) and lysosomes coordinate a network of key cellular processes including unfolded protein response (UPR) and autophagy in response to stress. How ER stress is signaled to lysosomes remains elusive. Here we find that ER disturbance activates chaperone-mediated autophagy (CMA). ER stressors lead to a PERK-dependent activation and recruitment of MKK4 to lysosomes, activating p38 MAPK at lysosomes. Lysosomal p38 MAPK directly phosphorylates the CMA receptor LAMP2A at T211 and T213, which causes its membrane accumulation and active conformational change, activating CMA. Loss of ER stress-induced CMA activation sensitizes cells to ER stress-induced death. Neurotoxins associated with Parkinson's disease fully engages ER-p38 MAPK-CMA pathway in the mouse brain and uncoupling it results in a greater loss of SNc dopaminergic neurons. This work identifies the coupling of ER and CMA as a critical regulatory axis fundamental for physiological and pathological stress response.
-
WB
-
Mus musculus (House mouse)
-
Cell Biology
In Nat Commun on 24 November 2017 by Li, W., Zhu, J., et al.
Fig.1.D

-
WB
-
Mus musculus (House mouse)
Collected and cropped from Nat Commun by CiteAb, provided under a CC-BY license
Image 1 of 5
In Nat Commun on 24 November 2017 by Li, W., Zhu, J., et al.
Fig.1.B

-
WB
-
Mus musculus (House mouse)
Collected and cropped from Nat Commun by CiteAb, provided under a CC-BY license
Image 1 of 5
In Nat Commun on 24 November 2017 by Li, W., Zhu, J., et al.
Fig.5.B

-
WB
-
Mus musculus (House mouse)
Collected and cropped from Nat Commun by CiteAb, provided under a CC-BY license
Image 1 of 5
In Nat Commun on 24 November 2017 by Li, W., Zhu, J., et al.
Fig.8.A

-
WB
-
Mus musculus (House mouse)
Collected and cropped from Nat Commun by CiteAb, provided under a CC-BY license
Image 1 of 5
In Cell Death Discov on 1 January 2015 by Hashemi, S., Salma, J., et al.
Fig.5.D

-
WB
-
Collected and cropped from Cell Death Discov by CiteAb, provided under a CC-BY license
Image 1 of 5