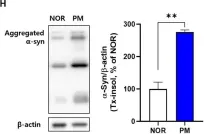


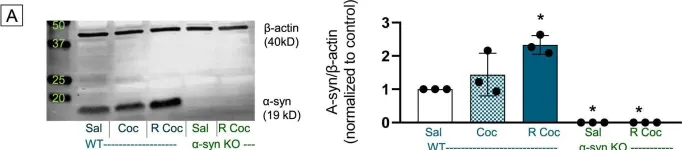
Parkinson's disease (PD) affects 1-2% of the global population and presents significant therapeutic challenges. Due to the limitations of existing treatments, there is a pressing need for alternative approaches. This study investigated the effects of invasive laser acupuncture (ILA), which combines acupuncture and photobiomodulation. In this method, optical fibers are inserted into the muscle layers of the acupoint to enhance therapeutic outcomes. Mice with MPTP-induced PD were treated with ILA at 830 nm or 650 nm. Protective effects of nigrostriatal dopaminergic neurons and fibers were assessed by examining TH immunoreactivity in the brain. Neuroinflammation markers in the brain and muscle metabolomic profiles were also analyzed. Comparisons between invasive and non-invasive laser application, as well as the impact of nerve blocking with lidocaine, were also evaluated. ILA at 830 nm (ILA830) significantly improved motor performance and increased the nigrostriatal TH-positive immunoreactivities. It reduced the levels of α-synuclein, apoptotic proteins, and inflammatory cytokines, while increasing anti-inflammatory in the brain. ILA830 also decreased nigrostriatal astrocyte and microglia activation. Muscle metabolomic analysis showed distinct group clustering and significant changes in metabolites like glucose and galactose, correlating with improved motor functions. Invasive laser treatment was more effective than non-invasive, and lidocaine pre-treatment did not block its effects. ILA at 830 nm effectively ameliorates PD symptoms by protecting dopaminergic neurons, and reducing neuroinflammation in the brain. Muscle metabolomic changes by ILA830, such as increased glucose and galactose, correlate with motor improvement. This approach offers a promising strategy for PD treatment, warranting further research to optimize its use in clinical settings.
© 2025. The Author(s).
Product Citations: 486
In Chinese Medicine on 7 May 2025 by Jeon, H., Oh, J. Y., et al.
-
Neuroscience
Synaptic vesicle-omics in mice captures signatures of aging and synucleinopathy.
In Nature Communications on 1 May 2025 by Gao, V., Chlebowicz, J., et al.
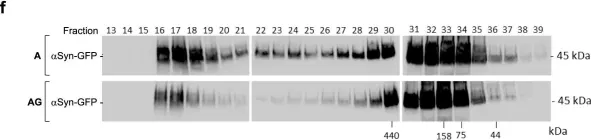
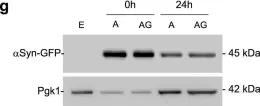
Neurotransmitter release occurs through exocytosis of synaptic vesicles. α-Synuclein's function and dysfunction in Parkinson's disease and other synucleinopathies is thought to be tightly linked to synaptic vesicle binding. Age is the biggest risk factor for synucleinopathy, and ~15% of synaptic vesicle proteins have been linked to central nervous system diseases. Yet, age- and disease-induced changes in synaptic vesicles remain unexplored. Via systematic analysis of synaptic vesicles at the ultrastructural, protein, and lipid levels, we reveal specific changes in synaptic vesicle populations, proteins, and lipids over age in wild-type mice and in α-synuclein knockout mice with and without expression of human α-synuclein. Strikingly, we find several previously undescribed synaptic changes in mice lacking α-synuclein, suggesting that loss of α-synuclein function contributes to synaptic dysfunction. These findings not only provide insights into synaptic vesicle biology and disease mechanisms in synucleinopathy, but also serve as a baseline for further mechanistic exploration of age- and disease-related alterations in synaptic vesicles.
© 2025. The Author(s).
Microglia-driven inflammation induces progressive tauopathies and synucleinopathies.
In Experimental & Molecular Medicine on 1 May 2025 by Lee, S. H., Bae, E. J., et al.
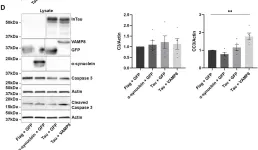
Alzheimer's disease and Parkinson's disease are characterized by distinct types of abnormal protein aggregates within neurons. These aggregates are known as neurofibrillary tangles and Lewy bodies, which consist of tau and α-synuclein, respectively. As the diseases progress, these aggregates spread from one cell to another, causing protein pathology to affect broader regions of the brain. Another notable characteristic of these diseases is neuroinflammation, which occurs when microglia become activated. Recent studies have suggested that inflammation may contribute to the formation and propagation of protein aggregates. However, it remains unclear whether microglia-driven inflammation can initiate and propagate different proteinopathies and associated neuropathology in neurodegenerative diseases. Here, using single-cell RNA sequencing, we observed that microglia exposed to α-synuclein or tau underwent changes in their characteristics and displayed distinct types of inflammatory response. The naive mice that received these microglial cell transplants developed both tauopathy and synucleinopathy, along with gliosis and inflammation. Importantly, these pathological features were not limited to the injection sites but also spread to other regions of the brain, including the opposite hemisphere. In conjunction with these pathological changes, the mice experienced progressive motor and cognitive deficits. These findings conclusively demonstrate that microglia-driven inflammation alone can trigger the full range of pathological features observed in neurodegenerative diseases, and that inflammation-induced local neuropathology can spread to larger brain regions. Consequently, these results suggest that microglia-driven inflammation plays an early and pivotal role in the development of neurodegenerative diseases. The transplantation of microglia activated by αSyn or tau proteins into the brains of naive mice resulted in the formation of synucleinopathy, tauopathy, gliosis, neuroinflammation and behavioral abnormalities. Activated microglia displayed alterations in subclusters as well as the corresponding feature genes.
© 2025. The Author(s).
-
WB
-
Mus musculus (House mouse)
-
Biochemistry and Molecular biology
-
Immunology and Microbiology
-
Neuroscience
Alpha-synuclein phosphorylation is abundant in the non-synucleinopathy human brain
Preprint on BioRxiv : the Preprint Server for Biology on 26 April 2025 by Choi, S., Duvernay, J., et al.
Phosphorylation of alpha-synuclein at serine 129 (PS129) has been strongly associated with aggregates and implicated in the synucleinopathy disease processes. Recent findings suggest that PS129 is abundant in the non-synucleinopathy mammalian brain, in a brain region-specific and context-dependent manner, raising important questions about the significance of PS129 for synucleinopathy disease processes. Here, we identified and described phosphatase labile physiological PS129 in the mammalian brain using a range of specimens from rodent to human surgically resected temporal lobectomy tissues. Results confirmed phosphatase-sensitive physiological PS129 in the non-synucleinopathy human temporal lobe and hippocampus when the post-mortem interval was eliminated (i.e., surgically resected human samples or perfusion-fixed animal brain). In the non-synucleinopathy mammalian brain, PS129 fluctuated between 2-45% of the total asyn pool in a case-specific and brain region-specific manner. In agreement with previous studies, physiological PS129 was abundantly distributed throughout the layers of the hippocampus and cortex. Physiological PS129 was not observed in PD brain, or non-synucleinopathy specimens of >2h post-mortem interval. Remarkably, in the PD brain, the PS129 to asyn ratio was similar (∼5-40%) to that in the non-synucleinopathy brain. Despite physiological PS129 being associated with neuronal activity, physiological PS129 did not correlate with cFos expression. Conclusions: Physiological PS129 is abundant in the human brain, and apparent pathological enrichment, in part, can be explained by the enzymatic resistance of aggregates and not disease-driven phosphorylation events.
Liganded magnetic nanoparticles for magnetic resonance imaging of α-synuclein.
In NPJ Parkinson's Disease on 23 April 2025 by Pan, H., Balbirnie, M., et al.
Aggregation of the protein α-synuclein (α-syn) is the histopathological hallmark of neurodegenerative diseases such as Parkinson's disease (PD), dementia with Lewy bodies (DLB), and multiple system atrophy (MSA), which are collectively known as synucleinopathies. Currently, patients with synucleinopathies are diagnosed by physical examination and medical history, often at advanced stages of disease. Because synucleinopathies are associated with α-syn aggregates, and α-syn aggregation often precedes onset of symptoms, detecting α-syn aggregates would be a valuable early diagnostic for patients with synucleinopathies. Here, we design a liganded magnetic nanoparticle (LMNP) functionalized with an α-syn-targeting peptide to be used as a magnetic resonance imaging (MRI)-based biomarker for α-syn. Our LMNPs bind to aggregates of α-syn in vitro, cross the blood-brain barrier in mice with mannitol adjuvant, and can be used as an MRI contrast agent to distinguish mice with α-synucleinopathy from age-matched, wild-type control mice in vivo. These results provide evidence for the potential of magnetic nanoparticles that target α-syn for diagnosis of synucleinopathies.
© 2025. The Author(s).
-
ELISA
-
Homo sapiens (Human)
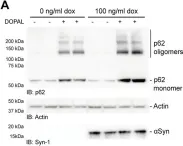
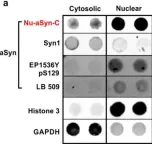
In Cell Death Dis on 18 June 2024 by Masato, A., Andolfo, A., et al.
Fig.7.A

-
WB
-
Collected and cropped from Cell Death Dis by CiteAb, provided under a CC-BY license
Image 1 of 59
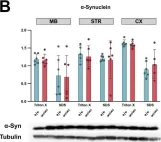
In MicroPubl Biol on 29 March 2024 by Dimopoulou, A., Panagiotakopoulou, V., et al.
Fig.1.B

-
WB
-
Mus musculus (House mouse)
Collected and cropped from MicroPubl Biol by CiteAb, provided under a CC-BY license
Image 1 of 59
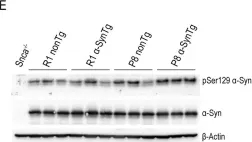
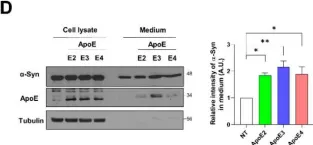
In Sci Rep on 30 January 2024 by Perez-Villalba, A., Sirerol-Piquer, M. S., et al.
Fig.4.E

-
WB
-
Mus musculus (House mouse)
Collected and cropped from Sci Rep by CiteAb, provided under a CC-BY license
Image 1 of 59
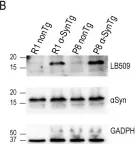
In Sci Rep on 30 January 2024 by Perez-Villalba, A., Sirerol-Piquer, M. S., et al.
Fig.1.B

-
WB
-
Mus musculus (House mouse)
Collected and cropped from Sci Rep by CiteAb, provided under a CC-BY license
Image 1 of 59
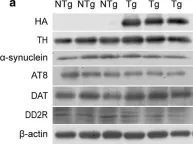
In Front Mol Neurosci on 28 August 2023 by Huh, E., Choi, J. G., et al.
Fig.1.H

-
WB
-
Collected and cropped from Front Mol Neurosci by CiteAb, provided under a CC-BY license
Image 1 of 59
In Nat Commun on 6 April 2023 by Rosado-Ramos, R., Poças, G. M., et al.
Fig.2.D

-
WB
-
Saccharomyces cerevisiae (Yeast)
Collected and cropped from Nat Commun by CiteAb, provided under a CC-BY license
Image 1 of 59
In Nat Commun on 6 April 2023 by Rosado-Ramos, R., Poças, G. M., et al.
Fig.2.E

-
WB
-
Saccharomyces cerevisiae (Yeast)
Collected and cropped from Nat Commun by CiteAb, provided under a CC-BY license
Image 1 of 59
In Nat Commun on 6 April 2023 by Rosado-Ramos, R., Poças, G. M., et al.
Fig.2.F

-
WB
-
Saccharomyces cerevisiae (Yeast)
Collected and cropped from Nat Commun by CiteAb, provided under a CC-BY license
Image 1 of 59
In Nat Commun on 6 April 2023 by Rosado-Ramos, R., Poças, G. M., et al.
Fig.2.G

-
WB
-
Saccharomyces cerevisiae (Yeast)
Collected and cropped from Nat Commun by CiteAb, provided under a CC-BY license
Image 1 of 59
In Front Cell Dev Biol on 1 November 2022 by Pilliod, J., Gélinas-Faucher, M., et al.
Fig.1.D

-
WB
-
Collected and cropped from Front Cell Dev Biol by CiteAb, provided under a CC-BY license
Image 1 of 59
In Biomolecules on 12 August 2022 by Pieger, K., Schmitt, V., et al.
Fig.3.A

-
WB
-
Collected and cropped from Biomolecules by CiteAb, provided under a CC-BY license
Image 1 of 59
In Int J Mol Sci on 27 July 2022 by Kang, S. J., Kim, S. J., et al.
Fig.5.D

-
WB
-
Collected and cropped from Int J Mol Sci by CiteAb, provided under a CC-BY license
Image 1 of 59
In Mol Brain on 1 December 2020 by Vanan, S., Zeng, X., et al.
Fig.6.A

-
WB
-
Mus musculus (House mouse)
Collected and cropped from Mol Brain by CiteAb, provided under a CC-BY license
Image 1 of 59
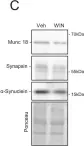
In Elife on 9 September 2020 by Monday, H. R., Bourdenx, M., et al.
Fig.4.C

-
WB
-
Collected and cropped from Elife by CiteAb, provided under a CC-BY license
Image 1 of 59
In Eneuro on 6 September 2020 by Verma, D. K., Ghosh, A., et al.
Fig.9.G

-
WB
-
Rattus norvegicus (Rat)
Collected and cropped from Eneuro by CiteAb, provided under a CC-BY license
Image 1 of 59
In Eneuro on 6 September 2020 by Verma, D. K., Ghosh, A., et al.
Fig.9.C

-
WB
-
Rattus norvegicus (Rat)
Collected and cropped from Eneuro by CiteAb, provided under a CC-BY license
Image 1 of 59
In Sci Rep on 30 July 2020 by Dominguez-Meijide, A., Vasili, E., et al.
Fig.2.A

-
WB
-
Homo sapiens (Human)
Collected and cropped from Sci Rep by CiteAb, provided under a CC-BY license
Image 1 of 59
In Invest Ophthalmol Vis Sci on 3 June 2020 by Kaehler, K., Seitter, H., et al.
Fig.1.A

-
WB
-
Mus musculus (House mouse)
Collected and cropped from Invest Ophthalmol Vis Sci by CiteAb, provided under a CC-BY license
Image 1 of 59
In Nat Commun on 13 March 2020 by Choi, I., Zhang, Y., et al.
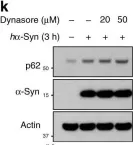
Fig.4.K

-
WB
-
Mus musculus (House mouse)
Collected and cropped from Nat Commun by CiteAb, provided under a CC-BY license
Image 1 of 59
In Nat Commun on 13 March 2020 by Choi, I., Zhang, Y., et al.
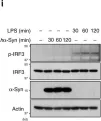
Fig.4.I

-
WB
-
Mus musculus (House mouse)
Collected and cropped from Nat Commun by CiteAb, provided under a CC-BY license
Image 1 of 59
In Nat Commun on 13 March 2020 by Choi, I., Zhang, Y., et al.
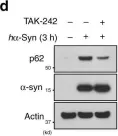
Fig.4.D

-
WB
-
Mus musculus (House mouse)
Collected and cropped from Nat Commun by CiteAb, provided under a CC-BY license
Image 1 of 59
In Nat Commun on 13 March 2020 by Choi, I., Zhang, Y., et al.
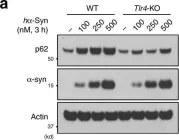
Fig.4.A

-
WB
-
Mus musculus (House mouse)
Collected and cropped from Nat Commun by CiteAb, provided under a CC-BY license
Image 1 of 59
In Commun Biol on 23 November 2019 by Trubetckaia, O., Lane, A. E., et al.
Fig.3.A

-
WB
-
Collected and cropped from Commun Biol by CiteAb, provided under a CC-BY license
Image 1 of 59
In Sci Rep on 21 May 2019 by Uehara, T., Choong, C. J., et al.
Fig.6.C

-
WB
-
Mus musculus (House mouse)
Collected and cropped from Sci Rep by CiteAb, provided under a CC-BY license
Image 1 of 59
In Transl Neurodegener on 15 March 2019 by Zou, W., Pu, T., et al.
Fig.6.C

-
WB
-
Mus musculus (House mouse)
Collected and cropped from Transl Neurodegener by CiteAb, provided under a CC-BY license
Image 1 of 59