
Targeting DNA repair proteins with small-molecule inhibitors became a proven anti-cancer strategy. Previously, we identified an inhibitor of a major protein of homologous recombination (HR) RAD51, named B02. B02 inhibited HR in human cells and sensitized them to chemotherapeutic drugs in vitro and in vivo. Here, using a medicinal chemistry approach, we aimed to improve the potency of B02. We identified the B02 analog, B02-isomer, which inhibits HR in human cells with significantly higher efficiency. We also show that B02-iso sensitizes triple-negative breast cancer MDA-MB-231 cells to the PARP inhibitor (PARPi) olaparib.
Product Citations: 11
New RAD51 Inhibitors to Target Homologous Recombination in Human Cells.
In Genes on 16 June 2021 by Shkundina, I. S., Gall, A. A., et al.
-
WB
-
Homo sapiens (Human)
-
Genetics
Show More
Show Less
In Journal of Cell Science on 20 May 2019 by Wang, Z., Feng, J., et al.
Necroptosis is a regulated form of necrotic cell death that is mediated by receptor-interacting serine/threonine-protein kinase 1 (RIPK1), RIPK3 and mixed-lineage kinase domain-like protein (MLKL), which mediates necroptotic signal transduction induced by tumor necrosis factor (TNF). Although many target proteins for necroptosis have been identified, no report had indicated that FK506-binding protein 12 (FKBP12, also known as FKBP1A), an endogenous protein that regulates protein folding and conformation alteration, is involved in mediating necroptosis. In this study, we found that FKBP12 acts as a novel target protein in mediating necroptosis and the related systemic inflammatory response syndrome triggered by TNF. The mechanistic study discovered that FKBP12 is essential for initiating necrosome formation and RIPK1-RIPK3-MLKL signaling pathway activation in response to TNF receptor 1 ligation. In addition, FKBP12 is indispensable for RIPK1 and RIPK3 expression and subsequent spontaneous phosphorylation, which are essential processes for initial necrosome formation and necroptotic signal transduction; therefore, FKBP12 may target RIPK1 and RIPK3 to mediate necroptosis in vitro and in vivo Collectively, our data demonstrate that FKBP12 could be a potential therapeutic target for the clinical treatment of necroptosis-associated diseases.
© 2019. Published by The Company of Biologists Ltd.
-
WB
-
Homo sapiens (Human)
-
Cell Biology
Show More
Show Less
Rapalogs can promote cancer cell stemness in vitro in a Galectin-1 and H-ras-dependent manner.
In Oncotarget on 4 July 2017 by Posada, I. M. D., Lectez, B., et al.
Currently several combination treatments of mTor- and Ras-pathway inhibitors are being tested in cancer therapy. While multiple feedback loops render these central signaling pathways robust, they complicate drug targeting.Here, we describe a novel H-ras specific feedback, which leads to an inadvertent rapalog induced activation of tumorigenicity in Ras transformed cells. We find that rapalogs specifically increase nanoscale clustering (nanoclustering) of oncogenic H-ras but not K-ras on the plasma membrane. This increases H-ras signaling output, promotes mammosphere numbers in a H-ras-dependent manner and tumor growth in ovo. Surprisingly, also other FKBP12 binders, but not mTor-inhibitors, robustly decrease FKBP12 levels after prolonged (>2 days) exposure. This leads to an upregulation of the nanocluster scaffold galectin-1 (Gal-1), which is responsible for the rapamycin-induced increase in H-ras nanoclustering and signaling output. We provide evidence that Gal-1 promotes stemness features in tumorigenic cells. Therefore, it may be necessary to block inadvertent induction of stemness traits in H-ras transformed cells by specific Gal-1 inhibitors that abrogate its effect on H-ras nanocluster. On a more general level, our findings may add an important mechanistic explanation to the pleiotropic physiological effects that are observed with rapalogs.
-
WB
-
Cancer Research
Show More
Show Less
In PLoS Pathogens on 1 August 2014 by Perng, Y. C., Campbell, J. A., et al.
In this study, we have identified a unique mechanism in which human cytomegalovirus (HCMV) protein pUL79 acts as an elongation factor to direct cellular RNA polymerase II for viral transcription during late times of infection. We and others previously reported that pUL79 and its homologues are required for viral transcript accumulation after viral DNA synthesis. We hypothesized that pUL79 represented a unique mechanism to regulate viral transcription at late times during HCMV infection. To test this hypothesis, we analyzed the proteome associated with pUL79 during virus infection by mass spectrometry. We identified both cellular transcriptional factors, including multiple RNA polymerase II (RNAP II) subunits, and novel viral transactivators, including pUL87 and pUL95, as protein binding partners of pUL79. Co-immunoprecipitation (co-IP) followed by immunoblot analysis confirmed the pUL79-RNAP II interaction, and this interaction was independent of any other viral proteins. Using a recombinant HCMV virus where pUL79 protein is conditionally regulated by a protein destabilization domain ddFKBP, we showed that this interaction did not alter the total levels of RNAP II or its recruitment to viral late promoters. Furthermore, pUL79 did not alter the phosphorylation profiles of the RNAP II C-terminal domain, which was critical for transcriptional regulation. Rather, a nuclear run-on assay indicated that, in the absence of pUL79, RNAP II failed to elongate and stalled on the viral DNA. pUL79-dependent RNAP II elongation was required for transcription from all three kinetic classes of viral genes (i.e. immediate-early, early, and late) at late times during virus infection. In contrast, host gene transcription during HCMV infection was independent of pUL79. In summary, we have identified a novel viral mechanism by which pUL79, and potentially other viral factors, regulates the rate of RNAP II transcription machinery on viral transcription during late stages of HCMV infection.
-
Biochemistry and Molecular biology
-
Genetics
-
Immunology and Microbiology
Show More
Show Less
In Nucleic Acids Research on 1 June 2013 by Bindra, R. S., Goglia, A. G., et al.
Double-strand break (DSB) repair pathways are critical for the maintenance of genomic integrity and the prevention of tumorigenesis in mammalian cells. Here, we present the development and validation of a novel assay to measure mutagenic non-homologous end-joining (NHEJ) repair in living cells, which is inversely related to canonical NHEJ and is based on the sequence-altering repair of a single site-specific DSB at an intrachromosomal locus. We have combined this mutagenic NHEJ assay with an established homologous recombination (HR) assay such that both pathways can be monitored simultaneously. In addition, we report the development of a ligand-responsive I-SceI protein, in which the timing and kinetics of DSB induction can be precisely controlled by regulating protein stability and cellular localization in cells. Using this system, we report that mutagenic NHEJ repair is suppressed in growth-arrested and serum-deprived cells, suggesting that end-joining activity in proliferating cells is more likely to be mutagenic. Collectively, the novel DSB repair assay and inducible I-SceI will be useful tools to further elucidate the complexities of NHEJ and HR repair.
-
WB
-
Homo sapiens (Human)
-
Biochemistry and Molecular biology
Show More
Show Less
In Oncotarget on 4 July 2017 by Posada, I. M. D., Lectez, B., et al.
Fig.3.C

-
WB
-
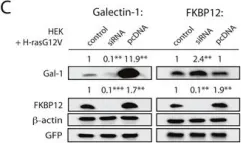
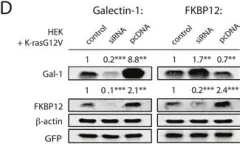
A galectin-1-dependent FKBP12 rescue-loop is activated upon rapalog-induced FKBP12 downmodulation(A and B) Nanoclustering-FRET analysis in FKBP12 knockdown/overexpressing HEK cells co-expressing mGFP- and mCherry-tagged (A) H-rasG12V or (B) K-rasG12V. The numbers in the bars indicate the number o...
more
A galectin-1-dependent FKBP12 rescue-loop is activated upon rapalog-induced FKBP12 downmodulation(A and B) Nanoclustering-FRET analysis in FKBP12 knockdown/overexpressing HEK cells co-expressing mGFP- and mCherry-tagged (A) H-rasG12V or (B) K-rasG12V. The numbers in the bars indicate the number of analyzed cells (mean ± SEM, n=4). (C and D) Western blot analysis in HEK cells transfected with the indicated FKBP12 or Gal-1 siRNAs, and in addition expressing (C) mGFP-H-rasG12V or (D) mGFP-K-rasG12V. Control is transfected with empty vector, while pcDNA marks exogenous expression of protein indicated on top of the blots. Numbers indicate β-actin normalized protein levels (n=4). (E and F) Western blot analysis of Ras and mTORC1 signaling in HEK cells expressing (E) mGFP-H-rasG12V or (F) mGFP-K-rasG12V under indicated FKBP12 or Gal-1 manipulations as in (C and D). Numbers indicate the ratio of phosphorylated to respective total protein levels (n=3).
less
Collected and cropped from Oncotarget by CiteAb, provided under a CC-BY license
Image 1 of 10
In Oncotarget on 4 July 2017 by Posada, I. M. D., Lectez, B., et al.
Fig.3.D

-
WB
-
A galectin-1-dependent FKBP12 rescue-loop is activated upon rapalog-induced FKBP12 downmodulation(A and B) Nanoclustering-FRET analysis in FKBP12 knockdown/overexpressing HEK cells co-expressing mGFP- and mCherry-tagged (A) H-rasG12V or (B) K-rasG12V. The numbers in the bars indicate the number o...
more
A galectin-1-dependent FKBP12 rescue-loop is activated upon rapalog-induced FKBP12 downmodulation(A and B) Nanoclustering-FRET analysis in FKBP12 knockdown/overexpressing HEK cells co-expressing mGFP- and mCherry-tagged (A) H-rasG12V or (B) K-rasG12V. The numbers in the bars indicate the number of analyzed cells (mean ± SEM, n=4). (C and D) Western blot analysis in HEK cells transfected with the indicated FKBP12 or Gal-1 siRNAs, and in addition expressing (C) mGFP-H-rasG12V or (D) mGFP-K-rasG12V. Control is transfected with empty vector, while pcDNA marks exogenous expression of protein indicated on top of the blots. Numbers indicate β-actin normalized protein levels (n=4). (E and F) Western blot analysis of Ras and mTORC1 signaling in HEK cells expressing (E) mGFP-H-rasG12V or (F) mGFP-K-rasG12V under indicated FKBP12 or Gal-1 manipulations as in (C and D). Numbers indicate the ratio of phosphorylated to respective total protein levels (n=3).
less
Collected and cropped from Oncotarget by CiteAb, provided under a CC-BY license
Image 1 of 10
In Oncotarget on 4 July 2017 by Posada, I. M. D., Lectez, B., et al.
Fig.6.C

-
WB
-
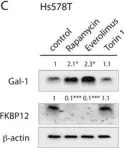
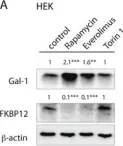
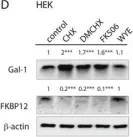
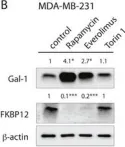
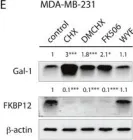
FKBP12-binding compounds stimulate FKBP12 downmodulation and couple Gal-1 induction(A to E) Western blot analysis of Gal-1 and FKBP12 protein levels in (A, D) HEK, (B, E) MDA-MB-231 and (C) Hs578T cells upon FKBP12 knockdown or treatment with 0.5 μM rapamycin, 2 μM everolimus, 0.25 μM torin 1, 0....
more
FKBP12-binding compounds stimulate FKBP12 downmodulation and couple Gal-1 induction(A to E) Western blot analysis of Gal-1 and FKBP12 protein levels in (A, D) HEK, (B, E) MDA-MB-231 and (C) Hs578T cells upon FKBP12 knockdown or treatment with 0.5 μM rapamycin, 2 μM everolimus, 0.25 μM torin 1, 0.18 μM CHX, 10 μM DM-CHX, 0.5 μM FK506 or 2 μM WYE -125132. Cells were treated for 48 h. Numbers indicate β-actin normalized protein levels (n=3).
less
Collected and cropped from Oncotarget by CiteAb, provided under a CC-BY license
Image 1 of 10
In Oncotarget on 4 July 2017 by Posada, I. M. D., Lectez, B., et al.
Fig.6.A

-
WB
-
FKBP12-binding compounds stimulate FKBP12 downmodulation and couple Gal-1 induction(A to E) Western blot analysis of Gal-1 and FKBP12 protein levels in (A, D) HEK, (B, E) MDA-MB-231 and (C) Hs578T cells upon FKBP12 knockdown or treatment with 0.5 μM rapamycin, 2 μM everolimus, 0.25 μM torin 1, 0....
more
FKBP12-binding compounds stimulate FKBP12 downmodulation and couple Gal-1 induction(A to E) Western blot analysis of Gal-1 and FKBP12 protein levels in (A, D) HEK, (B, E) MDA-MB-231 and (C) Hs578T cells upon FKBP12 knockdown or treatment with 0.5 μM rapamycin, 2 μM everolimus, 0.25 μM torin 1, 0.18 μM CHX, 10 μM DM-CHX, 0.5 μM FK506 or 2 μM WYE -125132. Cells were treated for 48 h. Numbers indicate β-actin normalized protein levels (n=3).
less
Collected and cropped from Oncotarget by CiteAb, provided under a CC-BY license
Image 1 of 10
In Oncotarget on 4 July 2017 by Posada, I. M. D., Lectez, B., et al.
Fig.6.D

-
WB
-
FKBP12-binding compounds stimulate FKBP12 downmodulation and couple Gal-1 induction(A to E) Western blot analysis of Gal-1 and FKBP12 protein levels in (A, D) HEK, (B, E) MDA-MB-231 and (C) Hs578T cells upon FKBP12 knockdown or treatment with 0.5 μM rapamycin, 2 μM everolimus, 0.25 μM torin 1, 0....
more
FKBP12-binding compounds stimulate FKBP12 downmodulation and couple Gal-1 induction(A to E) Western blot analysis of Gal-1 and FKBP12 protein levels in (A, D) HEK, (B, E) MDA-MB-231 and (C) Hs578T cells upon FKBP12 knockdown or treatment with 0.5 μM rapamycin, 2 μM everolimus, 0.25 μM torin 1, 0.18 μM CHX, 10 μM DM-CHX, 0.5 μM FK506 or 2 μM WYE -125132. Cells were treated for 48 h. Numbers indicate β-actin normalized protein levels (n=3).
less
Collected and cropped from Oncotarget by CiteAb, provided under a CC-BY license
Image 1 of 10
In Oncotarget on 4 July 2017 by Posada, I. M. D., Lectez, B., et al.
Fig.6.B

-
WB
-
FKBP12-binding compounds stimulate FKBP12 downmodulation and couple Gal-1 induction(A to E) Western blot analysis of Gal-1 and FKBP12 protein levels in (A, D) HEK, (B, E) MDA-MB-231 and (C) Hs578T cells upon FKBP12 knockdown or treatment with 0.5 μM rapamycin, 2 μM everolimus, 0.25 μM torin 1, 0....
more
FKBP12-binding compounds stimulate FKBP12 downmodulation and couple Gal-1 induction(A to E) Western blot analysis of Gal-1 and FKBP12 protein levels in (A, D) HEK, (B, E) MDA-MB-231 and (C) Hs578T cells upon FKBP12 knockdown or treatment with 0.5 μM rapamycin, 2 μM everolimus, 0.25 μM torin 1, 0.18 μM CHX, 10 μM DM-CHX, 0.5 μM FK506 or 2 μM WYE -125132. Cells were treated for 48 h. Numbers indicate β-actin normalized protein levels (n=3).
less
Collected and cropped from Oncotarget by CiteAb, provided under a CC-BY license
Image 1 of 10
In Oncotarget on 4 July 2017 by Posada, I. M. D., Lectez, B., et al.
Fig.6.E

-
WB
-
FKBP12-binding compounds stimulate FKBP12 downmodulation and couple Gal-1 induction(A to E) Western blot analysis of Gal-1 and FKBP12 protein levels in (A, D) HEK, (B, E) MDA-MB-231 and (C) Hs578T cells upon FKBP12 knockdown or treatment with 0.5 μM rapamycin, 2 μM everolimus, 0.25 μM torin 1, 0....
more
FKBP12-binding compounds stimulate FKBP12 downmodulation and couple Gal-1 induction(A to E) Western blot analysis of Gal-1 and FKBP12 protein levels in (A, D) HEK, (B, E) MDA-MB-231 and (C) Hs578T cells upon FKBP12 knockdown or treatment with 0.5 μM rapamycin, 2 μM everolimus, 0.25 μM torin 1, 0.18 μM CHX, 10 μM DM-CHX, 0.5 μM FK506 or 2 μM WYE -125132. Cells were treated for 48 h. Numbers indicate β-actin normalized protein levels (n=3).
less
Collected and cropped from Oncotarget by CiteAb, provided under a CC-BY license
Image 1 of 10
In Oncotarget on 4 July 2017 by Posada, I. M. D., Lectez, B., et al.
Fig.7.U

-
WB
-
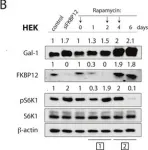
Rapamycin induces downmodulation of FKBP12 thus increasing galectin-1 expression(A) Proposed regulation scheme for Gal-1-dependent FKBP12-rescue loop. Low and High denotes concentration of proteins in the cell. Boxed numbers refer to stages in (B, C and D). Small arrows and block lines indicate m...
more
Rapamycin induces downmodulation of FKBP12 thus increasing galectin-1 expression(A) Proposed regulation scheme for Gal-1-dependent FKBP12-rescue loop. Low and High denotes concentration of proteins in the cell. Boxed numbers refer to stages in (B, C and D). Small arrows and block lines indicate modulators. (B to D) Western blot analysis of Gal-1 and FKBP12 protein levels in (B) HEK, (C) MDA-MB-231 and (D) Hs578T cells upon FKBP12 knockdown or rapamycin treatment for 6 days. Numbers indicate β-actin normalized protein levels. pS6K1 is shown as a marker of mTorC1 activity, and numbers indicate the ratio of phosphorylated to respective total protein levels (n=3). Arrows on top indicate when rapamycin treatment was refreshed. Boxed numbers on bottom relate to the steps in (A).
less
Collected and cropped from Oncotarget by CiteAb, provided under a CC-BY license
Image 1 of 10
In Oncotarget on 4 July 2017 by Posada, I. M. D., Lectez, B., et al.
Fig.7.B

-
WB
-
Rapamycin induces downmodulation of FKBP12 thus increasing galectin-1 expression(A) Proposed regulation scheme for Gal-1-dependent FKBP12-rescue loop. Low and High denotes concentration of proteins in the cell. Boxed numbers refer to stages in (B, C and D). Small arrows and block lines indicate m...
more
Rapamycin induces downmodulation of FKBP12 thus increasing galectin-1 expression(A) Proposed regulation scheme for Gal-1-dependent FKBP12-rescue loop. Low and High denotes concentration of proteins in the cell. Boxed numbers refer to stages in (B, C and D). Small arrows and block lines indicate modulators. (B to D) Western blot analysis of Gal-1 and FKBP12 protein levels in (B) HEK, (C) MDA-MB-231 and (D) Hs578T cells upon FKBP12 knockdown or rapamycin treatment for 6 days. Numbers indicate β-actin normalized protein levels. pS6K1 is shown as a marker of mTorC1 activity, and numbers indicate the ratio of phosphorylated to respective total protein levels (n=3). Arrows on top indicate when rapamycin treatment was refreshed. Boxed numbers on bottom relate to the steps in (A).
less
Collected and cropped from Oncotarget by CiteAb, provided under a CC-BY license
Image 1 of 10
In Oncotarget on 4 July 2017 by Posada, I. M. D., Lectez, B., et al.
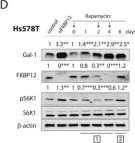
Fig.7.D

-
WB
-
Rapamycin induces downmodulation of FKBP12 thus increasing galectin-1 expression(A) Proposed regulation scheme for Gal-1-dependent FKBP12-rescue loop. Low and High denotes concentration of proteins in the cell. Boxed numbers refer to stages in (B, C and D). Small arrows and block lines indicate m...
more
Rapamycin induces downmodulation of FKBP12 thus increasing galectin-1 expression(A) Proposed regulation scheme for Gal-1-dependent FKBP12-rescue loop. Low and High denotes concentration of proteins in the cell. Boxed numbers refer to stages in (B, C and D). Small arrows and block lines indicate modulators. (B to D) Western blot analysis of Gal-1 and FKBP12 protein levels in (B) HEK, (C) MDA-MB-231 and (D) Hs578T cells upon FKBP12 knockdown or rapamycin treatment for 6 days. Numbers indicate β-actin normalized protein levels. pS6K1 is shown as a marker of mTorC1 activity, and numbers indicate the ratio of phosphorylated to respective total protein levels (n=3). Arrows on top indicate when rapamycin treatment was refreshed. Boxed numbers on bottom relate to the steps in (A).
less
Collected and cropped from Oncotarget by CiteAb, provided under a CC-BY license
Image 1 of 10