
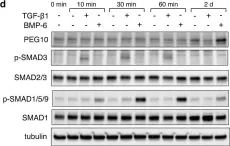
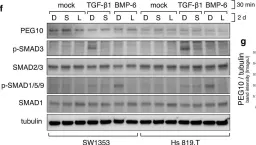
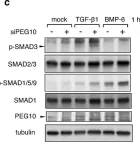
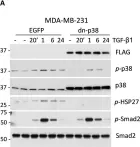



The transforming growth factor β (TGF-β) signaling superfamily includes NODAL and bone morphogenetic protein (BMP) signaling, which lead to the phosphorylation of different SMAD proteins and regulate key developmental events. Here, we present a protocol for immunofluorescence detection of phosphorylated SMAD proteins combined with other transcription factors in pre-implantation human embryos. We describe steps for segmenting the nuclei in human blastocysts and quantifying their immunofluorescence intensity. This protocol can be adapted to investigate TGF-β superfamily signaling activity in other mammalian embryos or in vitro models of their development. For complete details on the use and execution of this protocol, please refer to Brumm et al.1.
Copyright © 2025 The Authors. Published by Elsevier Inc. All rights reserved.
Product Citations: 108
In STAR Protocols on 20 June 2025 by Fallesen, T. & Brumm, A. S.
Preprint on BioRxiv : the Preprint Server for Biology on 21 January 2025 by Wits, M., Gómez-Suárez, N., et al.
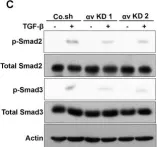
Background Fibrodysplasia ossificans progressiva (FOP) is caused by an activating mutation (p.R206H) in the type I BMP receptor ALK2, leading to heterotopic ossification (HO) in soft connective tissues. While aberrant Activin A-induced SMAD signaling is central in FOP pathogenesis, global signaling alterations remain poorly understood. Methods We performed phosphoproteomics, transcriptomics and biochemical analyses in mesenchymal cells (MSCs) overexpressing wild-type ALK2 WT or mutant ALK2 R206H receptors and in induced-MSCs derived from FOP patient iPSCs. Findings were validated in vivo using FOP-like mouse models and in vitro via pharmacological interventions. Results Multi-omics analyses revealed previously unrecognized signaling networks in ALK2 R206H cells, including enhanced MAPK, mTOR, RUNX2 and RHO-mediated mechanotransduction pathways. Notably, we identified dysregulated Activator Protein-1 (AP-1) expression and function as a novel contributor to FOP. AP-1 factors were highly enriched in HO lesions in FOP-like animals. Pharmacological inhibition of AP-1 significantly reduced osteochondrogenic differentiation in vitro. Conclusion This study highlights global signaling dysregulation in FOP and identifies AP-1 as a critical driver and potential therapeutic target for FOP.
In Cancer Gene Therapy on 1 December 2024 by Chen, Y. I., Tien, S. C., et al.
Tumor invasion is the hallmark of tumor malignancy. The invasive infiltration pattern of tumor cells located at the leading edge is highly correlated with metastasis and unfavorable patient outcomes. However, the regulatory mechanisms governing tumor malignancy at the invasive margin remain unclear. The IL-17B/IL-17RB pathway is known to promote pancreatic cancer invasion and metastasis, yet the specific mechanisms underlying IL-17RB upregulation during invasion are poorly understood. In this study, we unveiled a multistep process for IL-17RB upregulation at the invasive margin, which occurs through direct communication between tumor cells and fibroblasts. Tumor ATP1A1 facilitates plasma membrane expression of SEMA7A, which binds to and induces IGFBP-3 secretion from fibroblasts. The resulting gradient of IGFBP-3 influences the direction and enhances IL-17RB expression to regulate SNAI2 in invasion. These findings highlight the importance of local tumor-fibroblast interactions in promoting cancer cell invasiveness, potentially leading to the development of new therapeutic strategies targeting this communication.
© 2024. The Author(s).
-
WB
-
Cancer Research
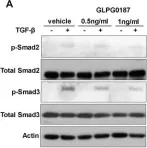
In Drug Development Research on 1 April 2024 by Kaya-Yasar, Y., Engin, S., et al.
The wingless/integrase-1 (WNT) pathway involved in the pathogenesis of inflammatory airway diseases has recently generated considerable research interest. Montelukast, a leukotriene receptor antagonist, provides therapeutic benefits in allergic asthma involving eosinophils. We aimed to investigate the role of the WNT pathway in the therapeutic actions of montelukast (MT) in a mixed type of allergic-acute airway inflammation model induced by ovalbumin (OVA) and lipopolysaccharide (LPS) in mice. Female mice were sensitized with intraperitoneal OVA-Al(OH)3 administration in the initiation phase and intranasal OVA followed by LPS administration in the challenge phase. The mice were divided into eight groups: control, asthmatic, and control/asthmatic treated with XAV939 (inhibitor of the canonical WNT pathway), LGK-974 (inhibitor of the secretion of WNT ligands), or MT at different doses. The inhibition of the WNT pathway prevented tracheal 5-HT and bradykinin hyperreactivity, while only the inhibition of the canonical WNT pathway partially reduced 5-HT and bradykinin contractions compared to the inflammation group. Therefore, MT treatment hindered 5-HT and bradykinin hyperreactivity associated with airway inflammation. Furthermore, MT prevented the increases in the phosphorylated GSK-3β and WNT5A levels, which had been induced by airway inflammation, in a dose-dependent manner. Conversely, the MT application caused a further increase in the fibronectin levels, while there was no significant alteration in the phosphorylation of the Smad-2 levels in the isolated lungs of the mice. The MT treatment reversed the increase in the mRNA expression levels of interleukin-17A. An increase in eosinophil and neutrophil counts was observed in bronchoalveolar lavage fluid samples obtained from the mice in the inflammation group, which was hampered by the MT treatment. The inhibition of the WNT pathway did not alter inflammatory cytokine expression or cell infiltration. The WNT pathway mediated the therapeutic effects of MT due to the inhibition of GSK-3β phosphorylation as well as the reduction of WNT5A levels in a murine airway inflammation model.
© 2024 Wiley Periodicals LLC.
-
WB
-
Immunology and Microbiology
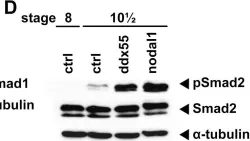
In Proceedings of the National Academy of Sciences of the United States of America on 22 August 2023 by van Dinther, M., Cunningham, K. T., et al.
Long-lived parasites evade host immunity through highly evolved molecular strategies. The murine intestinal helminth, Heligmosomoides polygyrus, down-modulates the host immune system through release of an immunosuppressive TGF-β mimic, TGM1, which is a divergent member of the CCP (Sushi) protein family. TGM1 comprises 5 domains, of which domains 1-3 (D1/2/3) bind mammalian TGF-β receptors, acting on T cells to induce Foxp3+ regulatory T cells; however, the roles of domains 4 and 5 (D4/5) remain unknown. We noted that truncated TGM1, lacking D4/5, showed reduced potency. Combination of D1/2/3 and D4/5 as separate proteins did not alter potency, suggesting that a physical linkage is required and that these domains do not deliver an independent signal. Coprecipitation from cells treated with biotinylated D4/5, followed by mass spectrometry, identified the cell surface protein CD44 as a coreceptor for TGM1. Both full-length and D4/5 bound strongly to a range of primary cells and cell lines, to a greater degree than D1/2/3 alone, although some cell lines did not respond to TGM1. Ectopic expression of CD44 in nonresponding cells conferred responsiveness, while genetic depletion of CD44 abolished enhancement by D4/5 and ablated the ability of full-length TGM1 to bind to cell surfaces. Moreover, CD44-deficient T cells showed attenuated induction of Foxp3 by full-length TGM1, to levels similar to those induced by D1/2/3. Hence, a parasite protein known to bind two host cytokine receptor subunits has evolved a third receptor specificity, which serves to raise the avidity and cell type-specific potency of TGF-β signaling in mammalian cells.
-
Immunology and Microbiology
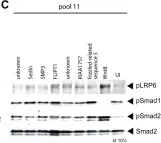
In Int J Mol Sci on 9 August 2023 by Hwang, S., Park, S., et al.
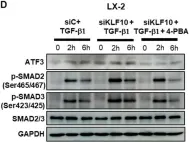
Fig.4.D

-
WB
-
Collected and cropped from Int J Mol Sci by CiteAb, provided under a CC-BY license
Image 1 of 26
In Int J Mol Sci on 9 August 2023 by Hwang, S., Park, S., et al.
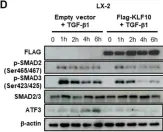
Fig.3.D

-
WB
-
Collected and cropped from Int J Mol Sci by CiteAb, provided under a CC-BY license
Image 1 of 26
In Int J Mol Sci on 9 August 2023 by Hwang, S., Park, S., et al.
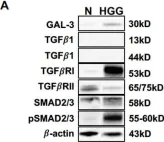
Fig.2.D

-
WB
-
Collected and cropped from Int J Mol Sci by CiteAb, provided under a CC-BY license
Image 1 of 26
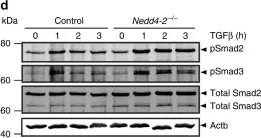
In Vet Sci on 19 June 2023 by Toedebusch, R. G., Wei, N. W., et al.
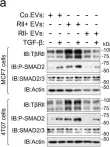
Fig.5.A

-
WB
-
Collected and cropped from Vet Sci by CiteAb, provided under a CC-BY license
Image 1 of 26
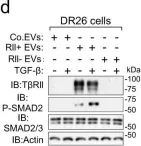
In Nat Commun on 1 August 2022 by Xie, F., Zhou, X., et al.
Fig.5.A

-
WB
-
Collected and cropped from Nat Commun by CiteAb, provided under a CC-BY license
Image 1 of 26
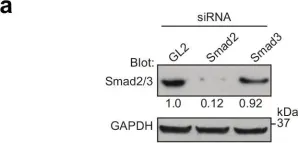
In Nat Commun on 1 August 2022 by Xie, F., Zhou, X., et al.
Fig.4.D

-
WB
-
Collected and cropped from Nat Commun by CiteAb, provided under a CC-BY license
Image 1 of 26
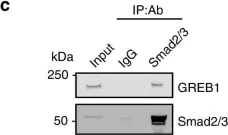
In Elife on 14 July 2022 by Shi, M., Tie, H. C., et al.
Fig.3.A

-
WB
-
Collected and cropped from Elife by CiteAb, provided under a CC-BY license
Image 1 of 26
In Nat Commun on 24 April 2020 by Duerr, J., Leitz, D. H. W., et al.
Fig.6.D

-
WB
-
Mus musculus (House mouse)
Collected and cropped from Nat Commun by CiteAb, provided under a CC-BY license
Image 1 of 26
In Nat Commun on 28 August 2019 by Matsumoto, S., Yamamichi, T., et al.
Fig.3.C

-
WB
-
Homo sapiens (Human)
Collected and cropped from Nat Commun by CiteAb, provided under a CC-BY license
Image 1 of 26
In Breast Cancer Res on 1 October 2018 by Zhang, H., Fredericks, T., et al.
Fig.6.A

-
WB
-
Collected and cropped from Breast Cancer Res by CiteAb, provided under a CC-BY license
Image 1 of 26
In Breast Cancer Res on 1 October 2018 by Zhang, H., Fredericks, T., et al.
Fig.6.C

-
WB
-
Collected and cropped from Breast Cancer Res by CiteAb, provided under a CC-BY license
Image 1 of 26
In Breast Cancer Res on 1 October 2018 by Zhang, H., Fredericks, T., et al.
Fig.6.E

-
WB
-
Collected and cropped from Breast Cancer Res by CiteAb, provided under a CC-BY license
Image 1 of 26
In Cell Death Dis on 1 May 2018 by Chen, L., Guo, P., et al.
Fig.2.E

-
WB
-
Collected and cropped from Cell Death Dis by CiteAb, provided under a CC-BY license
Image 1 of 26
In Sci Rep on 18 October 2017 by Shinohara, N., Maeda, S., et al.
Fig.4.D

-
WB
-
Collected and cropped from Sci Rep by CiteAb, provided under a CC-BY license
Image 1 of 26
In Sci Rep on 18 October 2017 by Shinohara, N., Maeda, S., et al.
Fig.4.F

-
WB
-
Collected and cropped from Sci Rep by CiteAb, provided under a CC-BY license
Image 1 of 26
In Sci Rep on 18 October 2017 by Shinohara, N., Maeda, S., et al.
Fig.5.C

-
WB
-
Collected and cropped from Sci Rep by CiteAb, provided under a CC-BY license
Image 1 of 26
In Oncotarget on 22 September 2017 by Limoge, M., Safina, A., et al.
Fig.1.A

-
WB
-
Collected and cropped from Oncotarget by CiteAb, provided under a CC-BY license
Image 1 of 26
In Nat Commun on 26 April 2017 by Xie, F., Jin, K., et al.
Fig.1.G

-
WB
-
Collected and cropped from Nat Commun by CiteAb, provided under a CC-BY license
Image 1 of 26
In Breast Cancer Res on 25 February 2015 by Li, Y., Drabsch, Y., et al.
Fig.4.C

-
WB
-
Collected and cropped from Breast Cancer Res by CiteAb, provided under a CC-BY license
Image 1 of 26
In Breast Cancer Res on 25 February 2015 by Li, Y., Drabsch, Y., et al.
Fig.5.A

-
WB
-
Collected and cropped from Breast Cancer Res by CiteAb, provided under a CC-BY license
Image 1 of 26
In PLoS One on 19 November 2013 by Zhang, S., Li, J., et al.
Fig.6.D

-
WB
-
Collected and cropped from PLoS One by CiteAb, provided under a CC-BY license
Image 1 of 26
In PLoS One on 19 November 2013 by Zhang, S., Li, J., et al.
Fig.4.C

-
WB
-
Collected and cropped from PLoS One by CiteAb, provided under a CC-BY license
Image 1 of 26
In PLoS One on 19 November 2013 by Zhang, S., Li, J., et al.
Fig.4.A

-
WB
-
Collected and cropped from PLoS One by CiteAb, provided under a CC-BY license
Image 1 of 26
In PLoS One on 19 November 2013 by Zhang, S., Li, J., et al.
Fig.4.B

-
WB
-
Collected and cropped from PLoS One by CiteAb, provided under a CC-BY license
Image 1 of 26
In PLoS One on 19 November 2013 by Zhang, S., Li, J., et al.
Fig.1.B

-
WB
-
Xenopus laevis (African clawed frog)
Collected and cropped from PLoS One by CiteAb, provided under a CC-BY license
Image 1 of 26