Resistance to chemotherapy is a major problem in the treatment of patients with triple-negative breast cancer (TNBC). Preclinical data suggest that TNBC is dependent on proteasomes; however, clinical observations indicate that the efficacy of proteasome inhibitors in TNBC may be limited, suggesting the need for combination therapies.
We compared bortezomib and carfilzomib and their combinations with nelfinavir and lopinavir in TNBC cell lines and primary cells with regard to their cytotoxic activity, functional proteasome inhibition, and induction of the unfolded protein response (UPR). Furthermore, we evaluated the involvement of sXBP1, ABCB1, and ABCG2 in the cytotoxic activity of drug combinations.
Carfilzomib, via proteasome β5 + β2 inhibition, is more cytotoxic in TNBC than bortezomib, which inhibits β5 + β1 proteasome subunits. The cytotoxicity of carfilzomib was significantly potentiated by nelfinavir or lopinavir. Carfilzomib with lopinavir induced endoplasmic reticulum stress and pro-apoptotic UPR through the accumulation of excess proteasomal substrate protein in TNBC in vitro. Moreover, lopinavir increased the intracellular availability of carfilzomib by inhibiting carfilzomib export from cells that express high levels and activity of ABCB1, but not ABCG2.
Proteasome inhibition by carfilzomib combined with nelfinavir/lopinavir represents a potential treatment option for TNBC, warranting further investigation.
© 2024. The Author(s).
Product Citations: 16
HIV-protease inhibitors potentiate the activity of carfilzomib in triple-negative breast cancer.
In British Journal of Cancer on 1 September 2024 by Besse, A., Sedlarikova, L., et al.
-
Homo sapiens (Human)
-
Cancer Research
In Scientific Reports on 17 March 2023 by Besse, L., Kraus, M., et al.
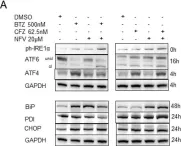
Chemotherapy resistance is still a major problem in the treatment of patients with non-small-cell-lung carcinoma (NSCLC), and novel concepts for the induction of cytotoxicity in NSCLC are highly warranted. Proteotoxicity, the induction of cytotoxicity by targeting the ubiquitin proteasome system, represents an appealing innovative strategy. The combination of the proteasome inhibitor bortezomib (BTZ) and the proteotoxic stress-inducing HIV drug nelfinavir (NFV) synergistically induces proteotoxicity and shows encouraging preclinical efficacy in NSCLC. The second-generation proteasome inhibitor carfilzomib (CFZ) is superior to BTZ and overcomes BTZ resistance in multiple myeloma patients. Here, we show that CFZ together with NFV is superior to the BTZ + NFV combination in inducing endoplasmic reticulum stress and proteotoxicity through the accumulation of excess proteasomal substrate protein in NSCLC in vitro and ex vivo. Interestingly, NFV increases the intracellular availability of CFZ through inhibition of CFZ export from NSCLC cells that express multidrug resistance (MDR) protein. Combining CFZ with NFV may therefore represent a future treatment option for NSCLC, which warrants further investigation.
© 2023. The Author(s).
-
WB
-
Homo sapiens (Human)
-
Cancer Research
Protein disulfide isomerase A1 as a novel redox sensor in VEGFR2 signaling and angiogenesis.
In Angiogenesis on 1 February 2023 by Nagarkoti, S., Kim, Y. M., et al.
VEGFR2 signaling in endothelial cells (ECs) is regulated by reactive oxygen species (ROS) derived from NADPH oxidases (NOXs) and mitochondria, which plays an important role in postnatal angiogenesis. However, it remains unclear how highly diffusible ROS signal enhances VEGFR2 signaling and reparative angiogenesis. Protein disulfide isomerase A1 (PDIA1) functions as an oxidoreductase depending on the redox environment. We hypothesized that PDIA1 functions as a redox sensor to enhance angiogenesis. Here we showed that PDIA1 co-immunoprecipitated with VEGFR2 or colocalized with either VEGFR2 or an early endosome marker Rab5 at the perinuclear region upon stimulation of human ECs with VEGF. PDIA1 silencing significantly reduced VEGF-induced EC migration, proliferation and spheroid sprouting via inhibiting VEGFR2 signaling. Mechanistically, VEGF stimulation rapidly increased Cys-OH formation of PDIA1 via the NOX4-mitochondrial ROS axis. Overexpression of "redox-dead" mutant PDIA1 with replacement of the active four Cys residues with Ser significantly inhibited VEGF-induced PDIA1-CysOH formation and angiogenic responses via reducing VEGFR2 phosphorylation. Pdia1+/- mice showed impaired angiogenesis in developmental retina and Matrigel plug models as well as ex vivo aortic ring sprouting model. Study using hindlimb ischemia model revealed that PDIA1 expression was markedly increased in angiogenic ECs of ischemic muscles, and that ischemia-induced limb perfusion recovery and neovascularization were impaired in EC-specific Pdia1 conditional knockout mice. These results suggest that PDIA1 can sense VEGF-induced H2O2 signal via CysOH formation to promote VEGFR2 signaling and angiogenesis in ECs, thereby enhancing postnatal angiogenesis. The oxidized PDIA1 is a potential therapeutic target for treatment of ischemic vascular diseases.
© 2022. The Author(s), under exclusive licence to Springer Nature B.V.
-
Cardiovascular biology
Late domain dependent E-cadherin recruitment into extracellular vesicles.
In Frontiers in Cell and Developmental Biology on 30 September 2022 by Bänfer, S., Kutscher, S., et al.
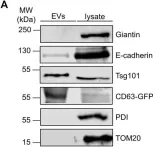
E-cadherin, a transmembrane protein involved in epithelial cell-cell adhesion and signaling, is found in exosomal fractions isolated from human body fluids. A cellular mechanism for recruitment of E-cadherin into extracellular vesicles (EVs) has not yet been defined. Here, we show that E-cadherin is incorporated into the membrane of EVs with the extracellular domain exposed at the vesicle surface. This recruitment depends on the endosomal sorting complex required for transport I (ESCRT-I) component Tsg101 and a highly conserved tetrapeptide P(S/T)AP late domain motif in the cytoplasmic tail of E-cadherin that mediates interaction with Tsg101. Mutation of this motif results in a loss of interaction and a dramatic decrease in exosomal E-cadherin secretion. We conclude, that the process of late domain mediated exosomal recruitment is exerted by this endogenous non-ESCRT transmembrane protein.
Copyright © 2022 Bänfer, Kutscher, Fleck, Dienst, Preußer, Pogge von Strandmann and Jacob.
-
WB
-
Canis lupus familiaris (Domestic dog)
In Nature Neuroscience on 1 June 2022 by Lee, J. H., Yang, D. S., et al.
Autophagy is markedly impaired in Alzheimer's disease (AD). Here we reveal unique autophagy dysregulation within neurons in five AD mouse models in vivo and identify its basis using a neuron-specific transgenic mRFP-eGFP-LC3 probe of autophagy and pH, multiplex confocal imaging and correlative light electron microscopy. Autolysosome acidification declines in neurons well before extracellular amyloid deposition, associated with markedly lowered vATPase activity and build-up of Aβ/APP-βCTF selectively within enlarged de-acidified autolysosomes. In more compromised yet still intact neurons, profuse Aβ-positive autophagic vacuoles (AVs) pack into large membrane blebs forming flower-like perikaryal rosettes. This unique pattern, termed PANTHOS (poisonous anthos (flower)), is also present in AD brains. Additional AVs coalesce into peri-nuclear networks of membrane tubules where fibrillar β-amyloid accumulates intraluminally. Lysosomal membrane permeabilization, cathepsin release and lysosomal cell death ensue, accompanied by microglial invasion. Quantitative analyses confirm that individual neurons exhibiting PANTHOS are the principal source of senile plaques in amyloid precursor protein AD models.
© 2022. The Author(s).
-
Neuroscience
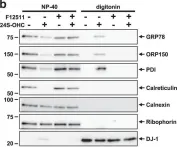
In Sci Rep on 17 March 2023 by Besse, L., Kraus, M., et al.
Fig.3.A

-
WB
-
Collected and cropped from Sci Rep by CiteAb, provided under a CC-BY license
Image 1 of 11
In Front Cell Dev Biol on 30 September 2022 by Bänfer, S., Kutscher, S., et al.
Fig.3.A

-
WB
-
Canis lupus familiaris (Domestic dog)
Collected and cropped from Front Cell Dev Biol by CiteAb, provided under a CC-BY license
Image 1 of 11
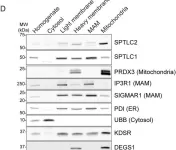
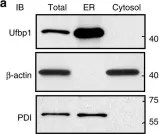
In Life Sci Alliance on 1 February 2022 by Aaltonen, M. J., Alecu, I., et al.
Fig.1.D

-
WB
-
Collected and cropped from Life Sci Alliance by CiteAb, provided under a CC-BY license
Image 1 of 11
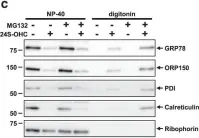
In Cell Death Discov on 10 July 2019 by Urano, Y., Vo, D. H., et al.
Fig.4.C

-
WB
-
Homo sapiens (Human)
Collected and cropped from Cell Death Discov by CiteAb, provided under a CC-BY license
Image 1 of 11
In Cell Death Discov on 10 July 2019 by Urano, Y., Vo, D. H., et al.
Fig.4.B

-
WB
-
Homo sapiens (Human)
Collected and cropped from Cell Death Discov by CiteAb, provided under a CC-BY license
Image 1 of 11
In Nat Commun on 6 March 2019 by Zhu, H., Bhatt, B., et al.
Fig.4.A

-
WB
-
Mus musculus (House mouse)
Collected and cropped from Nat Commun by CiteAb, provided under a CC-BY license
Image 1 of 11
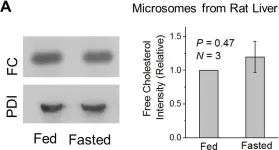
In PLoS One on 12 August 2017 by Sadh, K., Rai, P., et al.
Fig.3.A

-
WB
-
Rattus norvegicus (Rat)
Collected and cropped from PLoS One by CiteAb, provided under a CC-BY license
Image 1 of 11
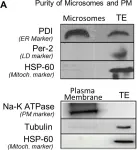
In PLoS One on 12 August 2017 by Sadh, K., Rai, P., et al.
Fig.2.A

-
WB
-
Rattus norvegicus (Rat)
Collected and cropped from PLoS One by CiteAb, provided under a CC-BY license
Image 1 of 11
In PLoS One on 12 August 2017 by Sadh, K., Rai, P., et al.
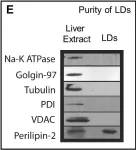
Fig.1.E

-
WB
-
Rattus norvegicus (Rat)
Collected and cropped from PLoS One by CiteAb, provided under a CC-BY license
Image 1 of 11
In PLoS One on 12 August 2017 by Sadh, K., Rai, P., et al.
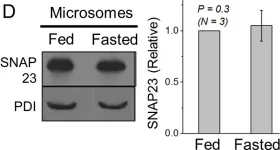
Fig.4.D

-
WB
-
Rattus norvegicus (Rat)
Collected and cropped from PLoS One by CiteAb, provided under a CC-BY license
Image 1 of 11
In PLoS One on 12 August 2017 by Sadh, K., Rai, P., et al.
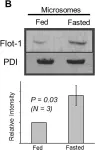
Fig.2.B

-
WB
-
Rattus norvegicus (Rat)
Collected and cropped from PLoS One by CiteAb, provided under a CC-BY license
Image 1 of 11