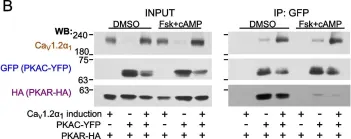
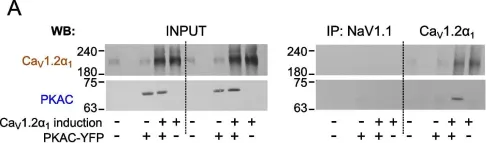
The β-adrenergic augmentation of cardiac contraction, by increasing the conductivity of L-type voltage-gated CaV1.2 channels, is of great physiological and pathophysiological importance. Stimulation of β-adrenergic receptors (βAR) activates protein kinase A (PKA) through separation of regulatory (PKAR) from catalytic (PKAC) subunits. Free PKAC phosphorylates the inhibitory protein Rad, leading to increased Ca2+ influx. In cardiomyocytes, the core subunit of CaV1.2, CaV1.2α1, exists in two forms: full-length or truncated (lacking the distal C-terminus (dCT)). Signaling efficiency is believed to emanate from protein interactions within multimolecular complexes, such as anchoring PKA (via PKAR) to CaV1.2α1 by A-kinase anchoring proteins (AKAPs). However, AKAPs are inessential for βAR regulation of CaV1.2 in heterologous models, and their role in cardiomyocytes also remains unclear.
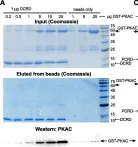
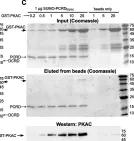
We show that PKAC interacts with CaV1.2α1 in heart and a heterologous model, independently of Rad, PKAR, or AKAPs. Studies with peptide array assays and purified recombinant proteins demonstrate direct binding of PKAC to two domains in CaV1.2α1-CT: the proximal and distal C-terminal regulatory domains (PCRD and DCRD), which also interact with each other. Data indicate both partial competition and possible simultaneous interaction of PCRD and DCRD with PKAC. The βAR regulation of CaV1.2α1 lacking dCT (which harbors DCRD) was preserved, but subtly altered, in a heterologous model, the Xenopus oocyte.
We discover direct interactions between PKAC and two domains in CaV1.2α1. We propose that these tripartite interactions, if present in vivo, may participate in organizing the multimolecular signaling complex and fine-tuning the βAR effect in cardiomyocytes.
© 2024. The Author(s).
Product Citations: 49
In BMC Biology on 28 November 2024 by Oz, S., Keren-Raifman, T., et al.
-
WB
-
Homo sapiens (Human)
-
Cardiovascular biology
GRK2 kinases in the primary cilium initiate SMOOTHENED-PKA signaling in the Hedgehog cascade.
In PLoS Biology on 1 August 2024 by Walker, M. F., Zhang, J., et al.
During Hedgehog (Hh) signal transduction in development and disease, the atypical G protein-coupled receptor (GPCR) SMOOTHENED (SMO) communicates with GLI transcription factors by binding the protein kinase A catalytic subunit (PKA-C) and physically blocking its enzymatic activity. Here, we show that GPCR kinase 2 (GRK2) orchestrates this process during endogenous mouse and zebrafish Hh pathway activation in the primary cilium. Upon SMO activation, GRK2 rapidly relocalizes from the ciliary base to the shaft, triggering SMO phosphorylation and PKA-C interaction. Reconstitution studies reveal that GRK2 phosphorylation enables active SMO to bind PKA-C directly. Lastly, the SMO-GRK2-PKA pathway underlies Hh signal transduction in a range of cellular and in vivo models. Thus, GRK2 phosphorylation of ciliary SMO and the ensuing PKA-C binding and inactivation are critical initiating events for the intracellular steps in Hh signaling. More broadly, our study suggests an expanded role for GRKs in enabling direct GPCR interactions with diverse intracellular effectors.
Copyright: © 2024 Walker et al. This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Preprint on BioRxiv : the Preprint Server for Biology on 28 May 2024 by Marsh, N. M., MacEwen, M. J. S., et al.
ABSTRACT Metabolic adaptations in response to changes in energy supply and demand are essential for survival. The mitochondrial calcium uniporter coordinates metabolic homeostasis by regulating TCA cycle activation, mitochondrial fatty acid oxidation and cellular calcium signaling. However, a comprehensive analysis of uniporter-regulated mitochondrial metabolic pathways has remained unexplored. Here, we investigate the metabolic consequences of uniporter loss- and gain-of-function, and identify a key transcriptional regulator that mediates these effects. Using gene expression profiling and proteomic, we find that loss of uniporter function increases the expression of proteins in the branched-chain amino acid (BCAA) catabolism pathway. Activity is further augmented through phosphorylation of the enzyme that catalyzes this pathway’s committed step. Conversely, in the liver cancer fibrolamellar carcinoma (FLC)-which we demonstrate to have high mitochondrial calcium levels-expression of BCAA catabolism enzymes is suppressed. We also observe uniporter-dependent suppression of the transcription factor KLF15, a master regulator of liver metabolic gene expression, including those involved in BCAA catabolism. Notably, loss of uniporter activity upregulates KLF15, along with its transcriptional target ornithine transcarbamylase (OTC), a component of the urea cycle, suggesting that uniporter hyperactivation may contribute to the hyperammonemia observed in FLC patients. Collectively, we establish that FLC has increased mitochondrial calcium levels, and identify an important role for mitochondrial calcium signaling in metabolic adaptation through the transcriptional regulation of metabolism.
-
WB
-
Cancer Research
-
Cell Biology
In Cardiovascular Research on 7 May 2024 by Shi, Q., Malik, H., et al.
A mechanistic link between depression and risk of arrhythmias could be attributed to altered catecholamine metabolism in the heart. Monoamine oxidase-A (MAO-A), a key enzyme involved in catecholamine metabolism and longstanding antidepressant target, is highly expressed in the myocardium. The present study aimed to elucidate the functional significance and underlying mechanisms of cardiac MAO-A in arrhythmogenesis.
Analysis of the TriNetX database revealed that depressed patients treated with MAO inhibitors had a lower risk of arrhythmias compared with those treated with selective serotonin reuptake inhibitors. This effect was phenocopied in mice with cardiomyocyte-specific MAO-A deficiency (cMAO-Adef), which showed a significant reduction in both incidence and duration of catecholamine stress-induced ventricular tachycardia compared with wild-type mice. Additionally, cMAO-Adef cardiomyocytes exhibited altered Ca2+ handling under catecholamine stimulation, with increased diastolic Ca2+ reuptake, reduced diastolic Ca2+ leak, and diminished systolic Ca2+ release. Mechanistically, cMAO-Adef hearts had reduced catecholamine levels under sympathetic stress, along with reduced levels of reactive oxygen species and protein carbonylation, leading to decreased oxidation of Type II PKA and CaMKII. These changes potentiated phospholamban (PLB) phosphorylation, thereby enhancing diastolic Ca2+ reuptake, while reducing ryanodine receptor 2 (RyR2) phosphorylation to decrease diastolic Ca2+ leak. Consequently, cMAO-Adef hearts exhibited lower diastolic Ca2+ levels and fewer arrhythmogenic Ca2+ waves during sympathetic overstimulation.
Cardiac MAO-A inhibition exerts an anti-arrhythmic effect by enhancing diastolic Ca2+ handling under catecholamine stress.
© The Author(s) 2024. Published by Oxford University Press on behalf of the European Society of Cardiology. All rights reserved. For permissions, please e-mail: journals.permissions@oup.com.
-
WB
-
Cardiovascular biology
Protein kinase A is a functional component of focal adhesions.
In The Journal of Biological Chemistry on 1 May 2024 by Kang, M., Senatore, A. J., et al.
Focal adhesions (FAs) form the junction between extracellular matrix (ECM)-bound integrins and the actin cytoskeleton and also transmit signals that regulate cell adhesion, cytoskeletal dynamics, and cell migration. While many of these signals are rooted in reversible tyrosine phosphorylation, phosphorylation of FA proteins on Ser/Thr residues is far more abundant yet its mechanisms and consequences are far less understood. The cAMP-dependent protein kinase (protein kinase A; PKA) has important roles in cell adhesion and cell migration and is both an effector and regulator of integrin-mediated adhesion to the ECM. Importantly, subcellular localization plays a critically important role in specifying PKA function. Here, we show that PKA is present in isolated FA-cytoskeleton complexes and active within FAs in live cells. Furthermore, using kinase-catalyzed biotinylation of isolated FA-cytoskeleton complexes, we identify 53 high-stringency candidate PKA substrates within FAs. From this list, we validate tensin-3 (Tns3)-a well-established molecular scaffold, regulator of cell migration, and a component of focal and fibrillar adhesions-as a novel direct substrate for PKA. These observations identify a new pathway for phospho-regulation of Tns3 and, importantly, establish a new and important niche for localized PKA signaling and thus provide a foundation for further investigation of the role of PKA in the regulation of FA dynamics and signaling.
Copyright © 2024 The Authors. Published by Elsevier Inc. All rights reserved.
-
Biochemistry and Molecular biology
In BMC Biol on 28 November 2024 by Oz, S., Keren-Raifman, T., et al.
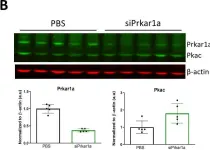
Fig.2.B

-
WB
-
Homo sapiens (Human)
Collected and cropped from BMC Biol by CiteAb, provided under a CC-BY license
Image 1 of 7
In BMC Biol on 28 November 2024 by Oz, S., Keren-Raifman, T., et al.
Fig.2.A

-
WB
-
Homo sapiens (Human)
Collected and cropped from BMC Biol by CiteAb, provided under a CC-BY license
Image 1 of 7
In BMC Biol on 28 November 2024 by Oz, S., Keren-Raifman, T., et al.
Fig.5.A

-
WB
-
Homo sapiens (Human)
Collected and cropped from BMC Biol by CiteAb, provided under a CC-BY license
Image 1 of 7
In BMC Biol on 28 November 2024 by Oz, S., Keren-Raifman, T., et al.
Fig.5.C

-
WB
-
Homo sapiens (Human)
Collected and cropped from BMC Biol by CiteAb, provided under a CC-BY license
Image 1 of 7
In PLoS One on 1 August 2020 by Soundarapandian, M. M., Juliana, C. A., et al.
Fig.1.B

-
WB
-
Collected and cropped from PLoS One by CiteAb, provided under a CC-BY license
Image 1 of 7
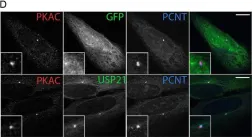
In J Cell Sci on 1 November 2016 by Heride, C., Rigden, D. J., et al.
Fig.6.D

-
ICC-IF
-
Collected and cropped from J Cell Sci by CiteAb, provided under a CC-BY license
Image 1 of 7
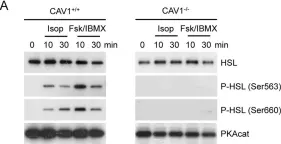
In PLoS One on 11 October 2012 by Martin, S., Fernandez-Rojo, M. A., et al.
Fig.1.A

-
WB
-
Mus musculus (House mouse)
Collected and cropped from PLoS One by CiteAb, provided under a CC-BY license
Image 1 of 7