Huntington’s disease (HD) is an autosomal dominant genetic neurodegenerative disease caused by a CAG expansion in the Huntingtin (HTT) gene, translating to an expanded polyglutamine tract in the huntingtin (HTT) protein. Age at disease onset is correlated to CAG repeat length, but varies by decades between individuals with identical repeat lengths. Genome-wide association studies link HD modification to DNA repair and mitochondrial health pathways. Recent clinical studies show elevated DNA damage in HD, even at the premanifest stage of disease. One of the major DNA repair nodes influencing neurodegenerative disease is the PARP pathway. Accumulation of poly ADP-ribose (PAR), produced by PARP1 and PARP2, has been implicated in the pathology of Alzheimer’s and Parkinson’s diseases, as well as autosomal recessive cerebellar ataxia. We report that HD mutation carriers have lower cerebrospinal fluid PAR levels than healthy controls, starting at the premanifest stage. Patient-derived fibroblasts have reduced PARP1/2 activity and elevated DNA damage, while elevated PAR levels are only revealed upon inhibition of PAR degradation. These phenotypes are rescued by moderate huntingtin level reduction via the huntingtin-lowering splice modulator drug, LMI070 (Branaplam). As a direct mechanism, we have defined a PAR-binding motif in huntingtin, detected huntingtin complexed with PARylated proteins in human cells during stress, and localized huntingtin to mitotic chromosomes upon inhibition of PAR degradation. Direct huntingtin PAR binding was measured by fluorescence polarization and visualized by atomic force microscopy. These results provide insight into a very early molecular mechanism of HD, suggesting possible targets in HD to design early preventive therapies.
Product Citations: 13
Poly ADP-Ribose Signaling is Dysregulated in Huntington’s Disease Patients
Preprint on BioRxiv : the Preprint Server for Biology on 24 November 2022 by Maiuri, T., Bazan, C. B., et al.
In International Journal of Molecular Sciences on 21 June 2022 by Jungwirth, J., Häring, C., et al.
The severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) causing the coronavirus disease-19 (COVID-19) is still challenging healthcare systems and societies worldwide. While vaccines are available, therapeutic strategies are developing and need to be adapted to each patient. Many clinical approaches focus on the repurposing of approved therapeutics against other diseases. However, the efficacy of these compounds on viral infection or even harmful secondary effects in the context of SARS-CoV-2 infection are sparsely investigated. Similarly, adverse effects of commonly used therapeutics against lifestyle diseases have not been studied in detail. Using mono cell culture systems and a more complex chip model, we investigated the effects of the acetylsalicylic acid (ASA) salt D,L-lysine-acetylsalicylate + glycine (LASAG) on SARS-CoV-2 infection in vitro. ASA is commonly known as Aspirin® and is one of the most frequently used medications worldwide. Our data indicate an inhibitory effect of LASAG on SARS-CoV-2 replication and SARS-CoV-2-induced expression of pro-inflammatory cytokines and coagulation factors. Remarkably, our data point to an additive effect of the combination of LASAG and the antiviral acting drug remdesivir on SARS-CoV-2 replication in vitro.
-
WB
-
Homo sapiens (Human)
-
COVID-19
ANKRD13a controls early cell-death checkpoint by interacting with RIP1 independent of NF-κB.
In Cell Death and Differentiation on 1 June 2022 by Won, M., Park, K. A., et al.
In TNF signaling, ubiquitination of RIP1 functions as an early cell-death checkpoint, which prevents the spatial transition of the signaling complex from complex-I to death-inducing complex-II. Here, we report that ankyrin repeat domain 13a (ANKRD13a) acts as a novel component of complex-II to set a higher signal threshold for the cytotoxic potential of TNF. ANKRD13a deficiency is sufficient to turn the response to TNF from survival to death by promoting the formation of complex-II without affecting NF-κB activation. ANKRD13a binds to ubiquitinated-RIP1 via its UIM, and subsequently limits the association of FADD and caspase-8 with RIP1. Moreover, high ANKRD13a expression is inversely correlated with apoptotic phenotypes in ovarian cancer tissues and is associated with poor prognosis. Our work identifies ANKRD13a as a novel gatekeeper of the early cell-death checkpoint, which may function as part of an escape mechanism from cell death in some cancers.
© 2021. The Author(s), under exclusive licence to ADMC Associazione Differenziamento e Morte Cellulare.
-
Cell Biology
PAAN/MIF nuclease inhibition prevents neurodegeneration in Parkinson's disease.
In Cell on 26 May 2022 by Park, H., Kam, T. I., et al.
Parthanatos-associated apoptosis-inducing factor (AIF) nuclease (PAAN), also known as macrophage migration inhibitor factor (MIF), is a member of the PD-D/E(X)K nucleases that acts as a final executioner in parthanatos. PAAN's role in Parkinson's disease (PD) and whether it is amenable to chemical inhibition is not known. Here, we show that neurodegeneration induced by pathologic α-synuclein (α-syn) occurs via PAAN/MIF nuclease activity. Genetic depletion of PAAN/MIF and a mutant lacking nuclease activity prevent the loss of dopaminergic neurons and behavioral deficits in the α-syn preformed fibril (PFF) mouse model of sporadic PD. Compound screening led to the identification of PAANIB-1, a brain-penetrant PAAN/MIF nuclease inhibitor that prevents neurodegeneration induced by α-syn PFF, AAV-α-syn overexpression, or MPTP intoxication in vivo. Our findings could have broad relevance in human pathologies where parthanatos plays a role in the development of cell death inhibitors targeting the druggable PAAN/MIF nuclease.
Copyright © 2022 Elsevier Inc. All rights reserved.
-
Neuroscience
In Redox Biology on 1 May 2021 by Yee, Y. H., Chong, S. J. F., et al.
Apart from its physiological role in inflammation and immunity, the nuclear factor-kappa B (NF-κB) protein complex has been implicated in tumorigenesis and its progression. Here, we provide evidence that a pro-oxidant milieu is an upstream effector of oncogenic NF-κB signaling. Through pharmacological or genetic inhibition of SOD1, we show that elevated intracellular superoxide (O2-) mediates sustained IKK phosphorylation, and induces downstream degradation of IκBα, leading to the nuclear localization and transcriptional activation of NF-κB. Mechanistically, we show that such sustained NF-κB signaling is a function of protein phosphatase 2A (PP2A) inactivation brought about by the nitrative modification of its substrate-binding sub-unit B56γ. Importantly, the pro-oxidant driven NF-κB activation enhances the migratory and invasive potential of cancer cells. In summary, our work highlights the critical involvement of O2--dependent peroxynitrite production in inhibiting PP2A-mediated dephosphorylation of IKK, thereby facilitating cancers to acquire an invasive phenotype. Given that NF-κB is a key player of chronic inflammation and carcinogenesis, our work unravels a novel synergistic node involving O2--driven redox milieu and deregulated PP2A as a potential therapeutic target.
Copyright © 2020 The Authors. Published by Elsevier B.V. All rights reserved.
-
WB
-
Homo sapiens (Human)
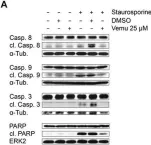
In Front Microbiol on 10 January 2018 by Holzberg, M., Boergeling, Y., et al.
Fig.3.A

-
WB
-
Homo sapiens (Human)
Collected and cropped from Front Microbiol by CiteAb, provided under a CC-BY license
Image 1 of 2
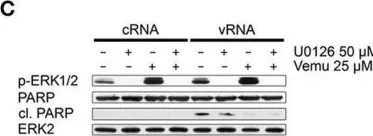
In Front Microbiol on 10 January 2018 by Holzberg, M., Boergeling, Y., et al.
Fig.3.C

-
WB
-
Homo sapiens (Human)
Collected and cropped from Front Microbiol by CiteAb, provided under a CC-BY license
Image 1 of 2