Maintenance of ploidy depends on the mitotic kinase Aurora B, the catalytic subunit of the chromosomal passenger complex (CPC) whose proficient activity is supported by HP1 enriched at inner centromeres. HP1 is known to associate with INCENP of the CPC in a manner that depends on the PVI motif conserved across HP1 interactors. Here, we found that the interaction of INCENP with HP1 requires not only the PVI motif but also its C-terminally juxtaposed domain. Remarkably, these domains conditionally fold the β-strand (PVI motif) and the α-helix from a disordered sequence upon HP1 binding and render INCENP with high affinity to HP1. This bipartite binding domain termed SSH domain (Structure composed of Strand and Helix) is necessary and sufficient to attain a predominant interaction of HP1 with INCENP. These results identify a unique HP1-binding module in INCENP that ensures enrichment of HP1 at inner centromeres, Aurora B activity, and thereby mitotic fidelity.
© 2024 Sako et al.
Product Citations: 52
Bipartite binding interface recruiting HP1 to chromosomal passenger complex at inner centromeres.
In The Journal of Cell Biology on 2 September 2024 by Sako, K., Furukawa, A., et al.
-
Cell Biology
-
Genetics
Topoisomerase I is an Evolutionarily Conserved Key Regulator for Satellite DNA Transcription
Preprint on BioRxiv : the Preprint Server for Biology on 5 May 2024 by Teng, Z., Yang, L., et al.
Repetitive satellite DNAs, divergent in nucleic-acid sequence and size across eukaryotes, provide a physical site for centromere assembly to orchestrate chromosome segregation during the cell cycle. These non-coding DNAs are transcribed by RNA polymerase (RNAP) II and the transcription has been shown to play a role in chromosome segregation, but a little is known about the regulation of centromeric transcription, especially in higher organisms with tandemly-repeated-DNA-sequence centromeres. Using RNA interference knockdown, chemical inhibition and AID/IAA degradation, we show that Topoisomerase I (TopI), not TopII, promotes the transcription of α-satellite DNAs, the main type of satellite on centromeres in human cells. Mechanistically, TopI localizes to centromeres, binds RNAP II and facilitates RNAP II elongation on centromeres. Interestingly, in response to DNA double-stranded breaks (DSBs) induced by chemotherapy drugs or CRSPR/Cas9, α-satellite transcription is dramatically stimulated in a DNA damage checkpoint-independent but TopI-dependent manner. These DSB-induced α-satellite RNAs were predominantly derived from the α-satellite high-order repeats of human centromeres and forms into strong speckles in the nucleus. Remarkably, TopI-dependent satellite transcription also exists in mouse 3T3 and Drosophila S2 cells and in Drosophila larval imaginal wing discs and tumor tissues. Altogether, our findings herein reveal an evolutionally conserved mechanism with TopI as a key player for the regulation of satellite transcription at both cellular and animal levels.
-
Biochemistry and Molecular biology
-
Genetics
Transient lagging chromosomes cause primary microcephaly
Preprint on BioRxiv : the Preprint Server for Biology on 2 May 2024 by Doria, E., Ivanova, D., et al.
ABSTRACT Primary microcephaly is caused by the depletion of neuronal progenitor cells during brain development, resulting in reduced brain size and impaired cognitive abilities. It arises due to recessive loss-of-function mutations of cell division genes, that are thought to cause neuronal progenitor loss either because of aneuploidy-driven apoptosis, spindle orientation defects, or prolonged mitotic timing. Loss of the two most frequently impaired microcephaly genes, WDR62 and ASPM , elicits, however, none of these phenotypes in human cells. Instead, their loss slows down poleward microtubule flux and results in transient lagging chromosomes in anaphase. Whether these defects cause primary microcephaly is, however, unknown. Here we show that slower poleward microtubule flux rates lead to transient lagging anaphase chromosomes that elicit an Aurora-B dependent activation of 53BP1 and the p53-target p21, impairing cell proliferation. Co-depletion of CAMSAP1, an inhibitor of microtubule depolymerization at spindle poles, restores normal poleward flux rates, suppresses the lagging chromosomes, silences 53BP1 and p21 activation, and allows normal cell proliferation in WDR62-depleted cells. In Drosophila melanogaster larvae knock-down of the CAMSAP1 ortholog Patronin suppresses the small brain, the neuroblast depletion, and the impaired cognitive phenotype associated with WDR62 loss. We thus postulate that poleward microtubule flux defects in neuronal progenitor cells drive primary microcephaly due to the activation of 53BP1 and p21 in response to transient lagging chromosomes in anaphase. Since loss of most cell division genes associated with primary microcephaly can lead to such transient lagging chromosomes, we speculate that they might represent a common cause of this disease.
-
Genetics
Mcph1, mutated in primary microcephaly, is also crucial for erythropoiesis.
In EMBO Reports on 1 May 2024 by Vial, Y., Nardelli, J., et al.
Microcephaly is a common feature in inherited bone marrow failure syndromes, prompting investigations into shared pathways between neurogenesis and hematopoiesis. To understand this association, we studied the role of the microcephaly gene Mcph1 in hematological development. Our research revealed that Mcph1-knockout mice exhibited congenital macrocytic anemia due to impaired terminal erythroid differentiation during fetal development. Anemia's cause is a failure to complete cell division, evident from tetraploid erythroid progenitors with DNA content exceeding 4n. Gene expression profiling demonstrated activation of the p53 pathway in Mcph1-deficient erythroid precursors, leading to overexpression of Cdkn1a/p21, a major mediator of p53-dependent cell cycle arrest. Surprisingly, fetal brain analysis revealed hypertrophied binucleated neuroprogenitors overexpressing p21 in Mcph1-knockout mice, indicating a shared pathophysiological mechanism underlying both erythroid and neurological defects. However, inactivating p53 in Mcph1-/- mice failed to reverse anemia and microcephaly, suggesting that p53 activation in Mcph1-deficient cells resulted from their proliferation defect rather than causing it. These findings shed new light on Mcph1's function in fetal hematopoietic development, emphasizing the impact of disrupted cell division on neurogenesis and erythropoiesis - a common limiting pathway.
© 2024. The Author(s).
-
Mus musculus (House mouse)
Sgo1 interacts with CENP-A to guide accurate chromosome segregation in mitosis.
In Journal of Molecular Cell Biology on 4 April 2024 by Wu, F., Akbar, H., et al.
Shugoshin-1 (Sgo1) is necessary for maintaining sister centromere cohesion and ensuring accurate chromosome segregation during mitosis. It has been reported that the localization of Sgo1 at the centromere is dependent on Bub1-mediated phosphorylation of histone H2A at T120. However, it remains uncertain whether other centromeric proteins play a role in regulating the localization and function of Sgo1 during mitosis. Here, we show that CENP-A interacts with Sgo1 and determines the localization of Sgo1 to the centromere during mitosis. Further biochemical characterization revealed that lysine and arginine residues in the C-terminal domain of Sgo1 are critical for binding CENP-A. Interestingly, the replacement of these basic amino acids with acidic amino acids perturbed the localization of Sgo1 and Aurora B to the centromere, resulting in aberrant chromosome segregation and premature chromatid separation. Taken together, these findings reveal a previously unrecognized but direct link between Sgo1 and CENP-A in centromere plasticity control and illustrate how the Sgo1-CENP-A interaction guides accurate cell division.
© The Author(s) (2023). Published by Oxford University Press on behalf of Journal of Molecular Cell Biology, CEMCS, CAS.
-
Cell Biology
-
Genetics
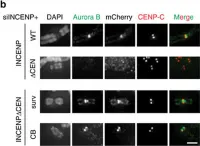
In Nat Commun on 31 May 2017 by Hengeveld, R. C. C., Vromans, M. J. M., et al.
Fig.1.B

-
ICC-IF
-
Homo sapiens (Human)
Collected and cropped from Nat Commun by CiteAb, provided under a CC-BY license
Image 1 of 2
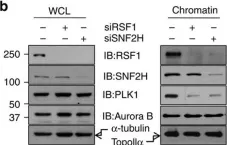
In Nat Commun on 10 August 2015 by Lee, H. S., Park, Y. Y., et al.
Fig.2.B

-
WB
-
Homo sapiens (Human)
Collected and cropped from Nat Commun by CiteAb, provided under a CC-BY license
Image 1 of 2