





Mitochondria are pivotal regulators of cellular homeostasis, integrating energy metabolism, biosynthesis, and programmed cell death (apoptosis). During apoptosis, mitochondrial outer membrane permeabilization by BCL-2-associated X protein/BCL-2 Homolog Antagonist Killer (BAX/BAK) pores facilitates release of apoptotic factors, while the role of inner mitochondrial membrane (IMM) remodeling remains less understood. Here, we identify serine beta-lactamase-like protein (LACTB), a filament-forming serine protease and tumor suppressor, as a regulator of IMM dynamics during apoptosis. LACTB suppression reduces cytochrome c release and apoptosis, whereas its overexpression promotes these effects. LACTB does not affect BAX or Drp1 recruitment to mitochondria. Rather, LACTB is required for apoptosis-induced mitochondrial remodeling, independent of OPA1 processing. Intriguingly, LACTB knockdown does not affect mitochondrial shape changes induced by CCCP treatment, suggesting that LACTB action is apoptosis-specific. Purified LACTB binds and remodels cardiolipin-enriched membrane nanotubes preferentially over planar lipid membranes, suggesting a direct effect in apoptotic membrane remodeling. Collectively, our findings suggest LACTB to be a mediator of apoptosis-induced IMM remodeling, a possible mechanism for tumor suppression in cancer.
Product Citations: 195
The tumor suppressor LACTB remodels mitochondria to promote cytochrome c release and apoptosis.
In Science Advances on 14 November 2025 by Kamerkar, S., Kang, T., et al.
-
Cancer Research
-
Cell Biology
UCP2 mediates mitochondrial dynamics to induce AgRP neuronal activity.
In Molecular Metabolism on 1 September 2025 by Jin, S., Yoon, N. A., et al.
The hypothalamic agouti-related protein (AgRP)- expressing neurons regulate feeding and whole-body energy homeostasis. A growing body of evidence indicates that changes in mitochondrial dynamics, such as fission and fusion, play a crucial role in regulating AgRP neuronal activity. However, the mechanisms underlying this process remain to be elucidated. Here, we showed a role of mitochondrial UCP2-mediated mitochondrial dynamics in AgRP neurons in regulating AgRP neuronal activity and fasting-induced feeding behavior.
We analyzed mitochondrial morphology, expression of activated dynamin-related protein 1 (DRP1), and mRNA expression levels of uncoupling protein 2 (Ucp2) in AgRP neurons of mice that were either in fed or fasted states. We then generated a mouse model in which Ucp2 was selectively deleted from adult AgRP neurons to assess the role of this mitochondrial protein in feeding behavior and whole-body energy metabolism.
We show fasting-induced AgRP neuronal activation is associated with UCP2-mediated mitochondrial fission and mitochondrial fatty acid utilization in AgRP neurons. In line with this, mice lacking UCP2 in AgRP neurons (Ucp2AgRPKO) show attenuated fasting- or ghrelin-induced AgRP neuronal activation and feeding behaviors and exhibited a significant decrease in body weight and fat mass accompanied by a significant increase in energy expenditure.
Altogether, our data revealed that UCP2-mediated mitochondrial dynamics and fatty acids oxidation in the hypothalamic AgRP neurons is necessary for AgRP neuronal function and fasting-induced food intake.
Copyright © 2025 The Authors. Published by Elsevier GmbH.. All rights reserved.
-
Biochemistry and Molecular biology
-
Cell Biology
In Biology on 20 July 2025 by Deshetty, U. M., Oladapo, A., et al.
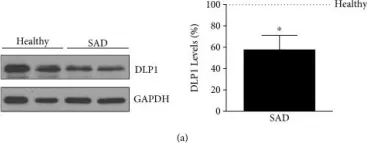
Cocaine misuse induces microglial activation and neuroinflammation, contributing to neurodegeneration and behavioral impairments. Prior studies have shown that cocaine induces mitochondrial dysfunction, dysregulated mitophagy, and lysosomal impairment in microglia. Here, we investigated the therapeutic potential of N-acetylcysteine (NAC) in mitigating cocaine-induced microglial activation and neuroinflammation. Mouse primary microglial cells (MPMs) were pretreated with NAC (5 mM) for 1 h prior to cocaine exposure (10 µM, 24 h) and analyzed for markers of microglial activation, mitophagy, and lysosomal integrity using Western blot, Seahorse assays, lysosomal pH, and membrane potential measurements. In vivo, C57BL/6N mice received NAC (200 mg/kg, i.p.) 1 h before daily cocaine injections (20 mg/kg, i.p.) for 7 days. Behavioral assays (open field, novel object recognition) and brain biomarker analyses (frontal cortex, hippocampus) were performed. Cocaine exposure elevated CD11b, mitophagy markers (PINK1, PARK, and DLP1), and autophagy proteins (Beclin1, and p62), while impairing mitochondrial and lysosomal functions. NAC pretreatment restored mitochondrial and lysosomal function, reduced reactive oxygen species, and normalized protein expression. In vivo, NAC also alleviated cocaine-induced microglial activation and behavioral deficits. These findings highlight NAC as a promising therapeutic agent to counteract cocaine-mediated neuroinflammation and neurotoxicity.
-
Neuroscience
Salt-derived lysosome impairment provokes tubular damage via mitochondrial quality control defects
Preprint on Research Square on 20 June 2025 by Taguchi, K., Kodama, G., et al.
Abstract Excessive salt consumption is associated with an increased risk of progressive kidney injury and heart failure. Understanding the mechanistic basis of high sodium (HS)-induced organ dysfunction is crucial for the development of novel pharmacotherapeutics. Herein, we show that exposure of kidney tubules to HS impairs lysosomal function via a reduction in V-ATPase expression, leading to dysregulation of mitophagy. HS also reduces mitofilin and DRP-1 in kidney tubular cells, thereby impairing mitochondrial dynamics, quality and function. Decreased renal levels of mitofilin are observed in patients with various kidney diseases and correlates with kidney dysfunction. Administration of mitochonic aid-5 (MA-5) preserves lysosomal function via restoration of V-ATPase and attenuates mitophagy, mitochondrial morphology and quality. MA-5 treatment also improves kidney and cardiac function in uninephrectomized mice administered deoxycorticosterone acetate with HS overload. Our present study suggests that MA-5 may play a protective role against HS-induced organ damages in the kidneys and hearts.
-
Cell Biology
Preprint on BioRxiv : the Preprint Server for Biology on 20 April 2025 by Koo, K., Choi, J., et al.
SUMMARY Mitochondrial function is critical for neural progenitor regulation, yet its dysregulation during early human brain development remains poorly defined. Megalencephalic leukoencephalopathy with subcortical cysts (MLC) is a neurodevelopmental disorder caused by MLC1 mutations, previously attributed to postnatal astrocyte dysfunction. Using patient-derived human cortical organoids, we show that MLC1 is expressed in early neuroepithelial cells. To assess mitochondrial state in live organoids, we developed the MAGO (Matrigel-coated gold nanostructure) platform for real-time, label-free detection of redox activity. MLC1 mutant organoids showed mitochondrial hyperactivation, increased ATP and ROS, reduced membrane potential, and altered fusion protein expression. These changes were accompanied by enhanced BrdU incorporation and expansion of PAX6⁺/SOX2⁺ progenitors. To assess the causal role of MLC1 mutation, we generated isogenic organoids using CRISPR prime editing, which recapitulated redox hyperactivation and increased proliferation. Our findings redefine MLC as a disorder of early mitochondrial and progenitor dysregulation and establish a tractable platform to study metabolic mechanisms in neurodevelopmental disease.
-
WB
-
Biochemistry and Molecular biology
-
Cell Biology
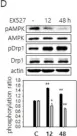
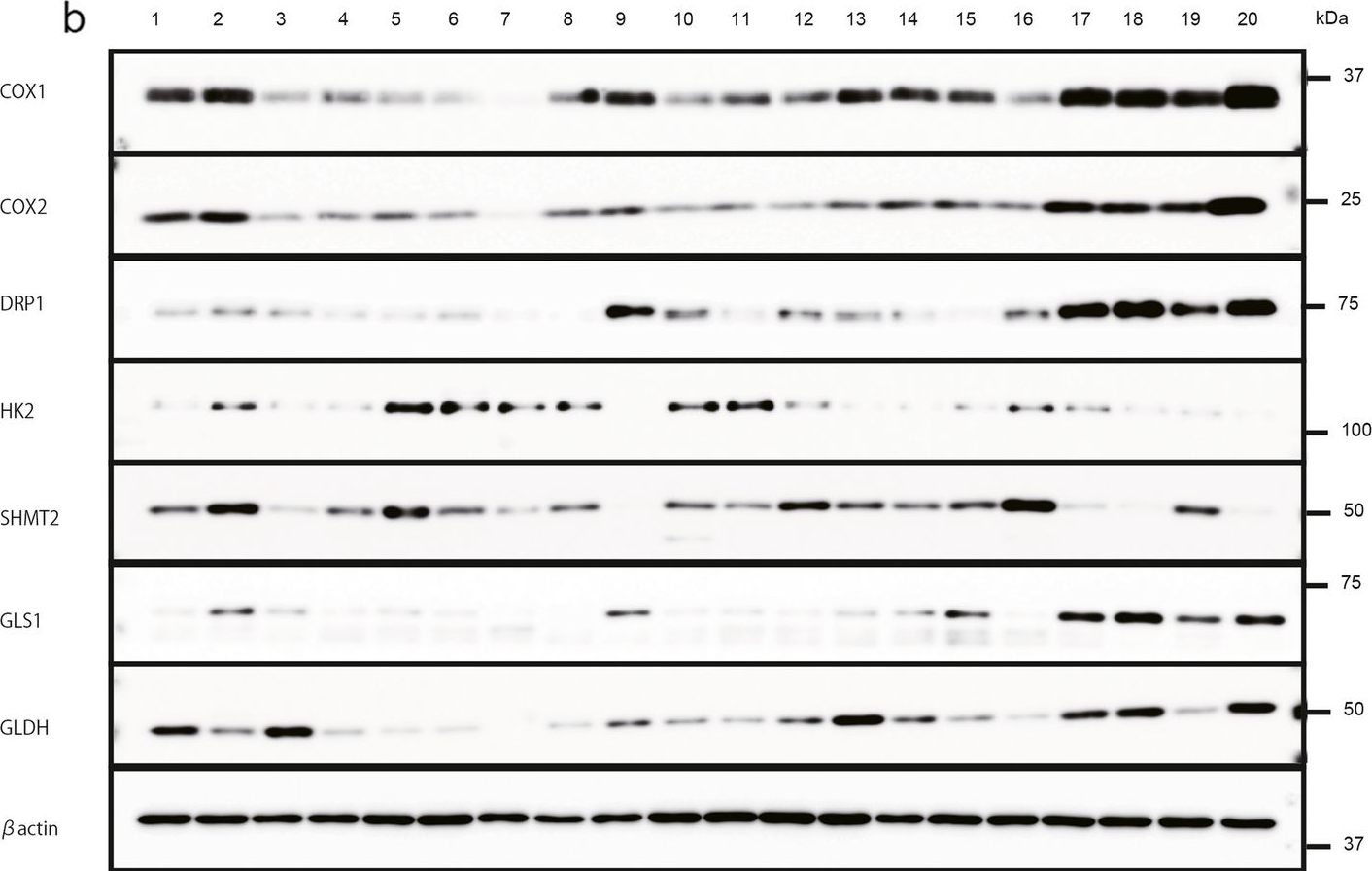
In Cancer Metab on 19 November 2024 by Miki, K., Yagi, M., et al.
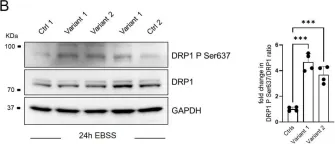
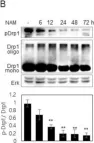
Fig.1.B
-
WB
-
Collected and cropped from Cancer Metabolism by CiteAb, provided under a CC-BY license
Image 1 of 53
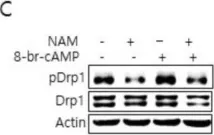
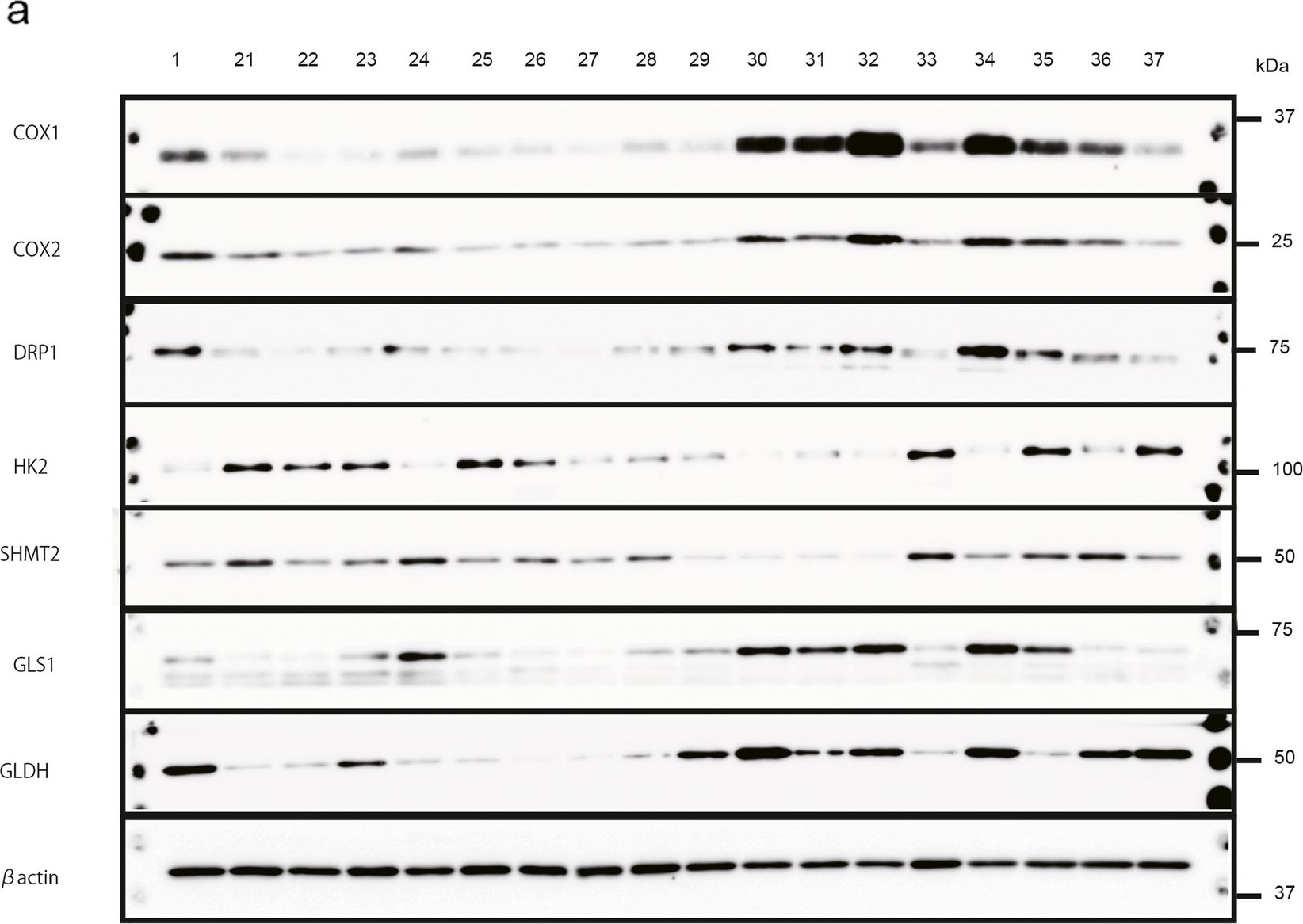
In Cancer Metab on 19 November 2024 by Miki, K., Yagi, M., et al.
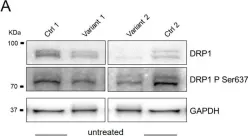
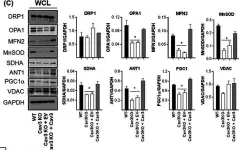
Fig.3.A
-
WB
-
Collected and cropped from Cancer Metabolism by CiteAb, provided under a CC-BY license
Image 1 of 53
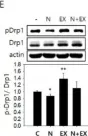
In Cells on 23 December 2023 by Wang, L., Rivas, R., et al.
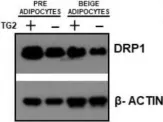
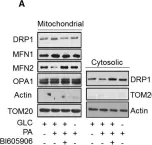
Fig.7.B

-
WB
-
Homo sapiens (Human)
Collected and cropped from Cells by CiteAb, provided under a CC-BY license
Image 1 of 53
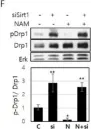
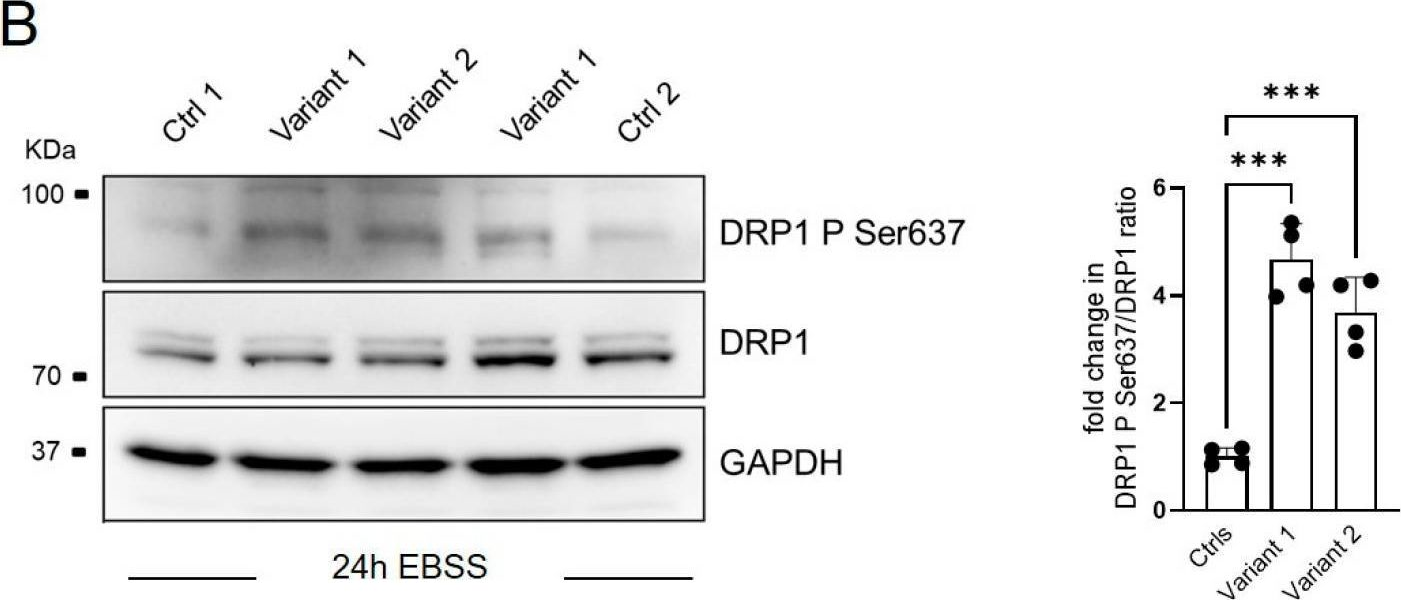
In Int J Mol Sci on 9 February 2023 by Naef, V., Meschini, M. C., et al.
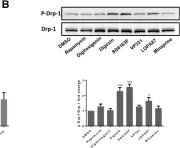
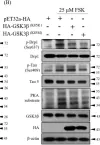
Fig.3.B
-
WB
-
Collected and cropped from International Journal of Molecular Sciences by CiteAb, provided under a CC-BY license
Image 1 of 53
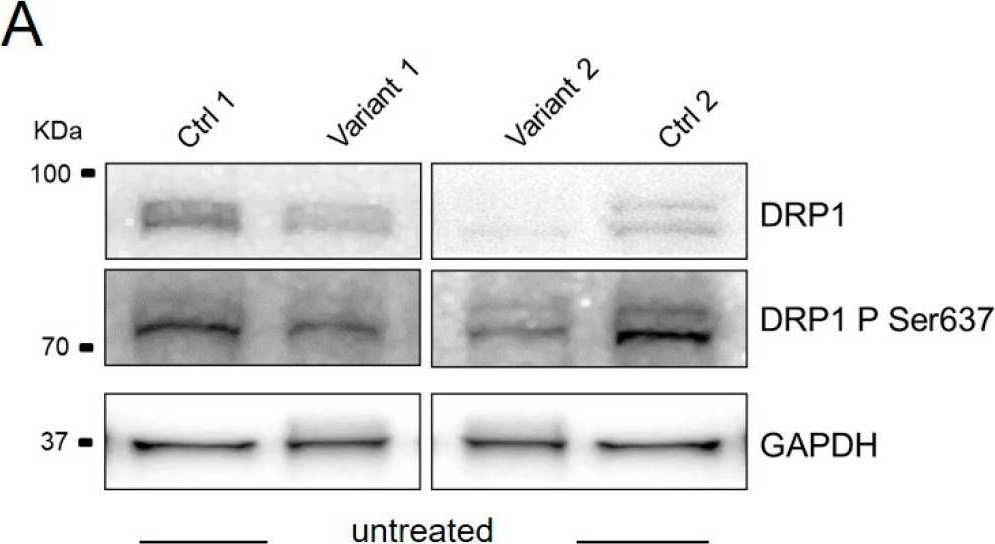
In Int J Mol Sci on 9 February 2023 by Naef, V., Meschini, M. C., et al.
Fig.3.A
-
WB
-
Collected and cropped from International Journal of Molecular Sciences by CiteAb, provided under a CC-BY license
Image 1 of 53
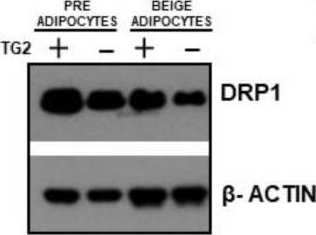
In Int J Mol Sci on 5 May 2022 by Lénárt, K., Banko, C., et al.
Fig.5.I
-
WB
-
Mus musculus (House mouse)
Collected and cropped from International Journal of Molecular Sciences by CiteAb, provided under a CC-BY license
Image 1 of 53
In Cells on 29 January 2022 by Liu, D., Peyre, F., et al.
Fig.7.B
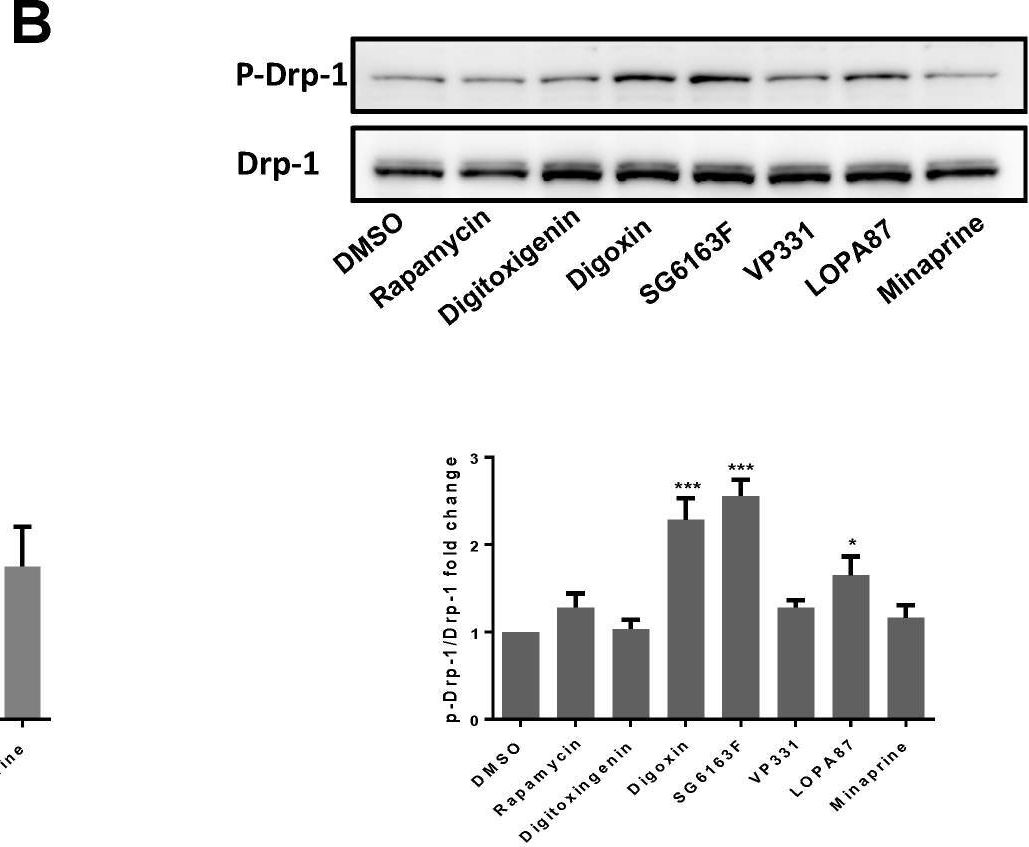
-
WB
-
Collected and cropped from Cells by CiteAb, provided under a CC-BY license
Image 1 of 53
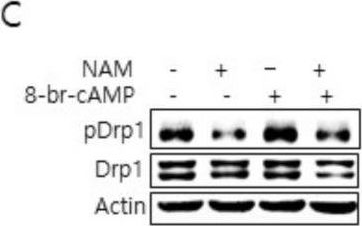
In Cells on 10 March 2021 by Song, S. B., Park, J. S., et al.
Fig.3.D
-
WB
-
Collected and cropped from Cells by CiteAb, provided under a CC-BY license
Image 1 of 53
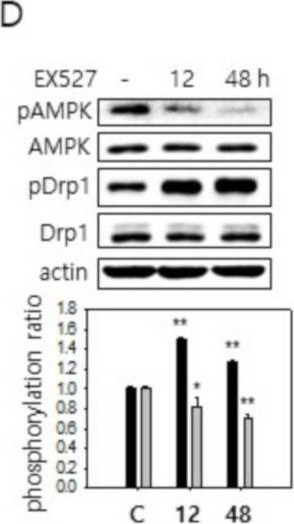
In Cells on 10 March 2021 by Song, S. B., Park, J. S., et al.
Fig.4.C
-
WB
-
Collected and cropped from Cells by CiteAb, provided under a CC-BY license
Image 1 of 53
In Cells on 10 March 2021 by Song, S. B., Park, J. S., et al.
Fig.3.L
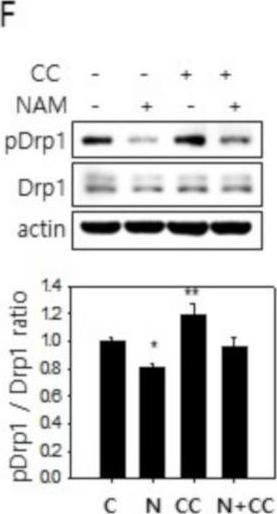
-
WB
-
Collected and cropped from Cells by CiteAb, provided under a CC-BY license
Image 1 of 53
In Cells on 10 March 2021 by Song, S. B., Park, J. S., et al.
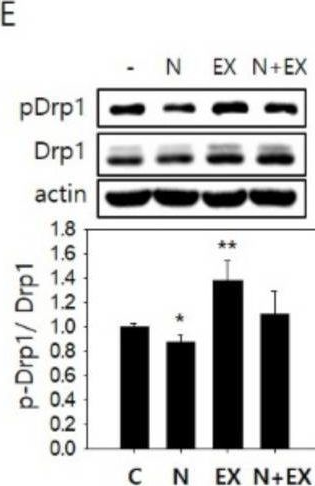
Fig.2.E
-
WB
-
Collected and cropped from Cells by CiteAb, provided under a CC-BY license
Image 1 of 53
In Cells on 10 March 2021 by Song, S. B., Park, J. S., et al.
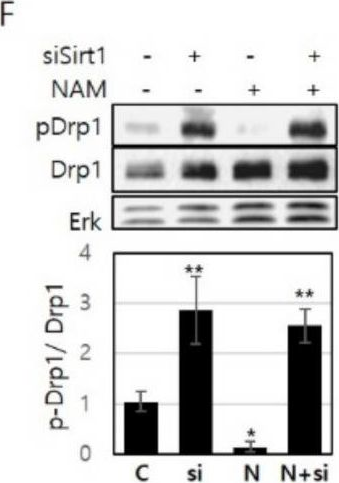
Fig.2.F
-
WB
-
Collected and cropped from Cells by CiteAb, provided under a CC-BY license
Image 1 of 53
In Cells on 10 March 2021 by Song, S. B., Park, J. S., et al.
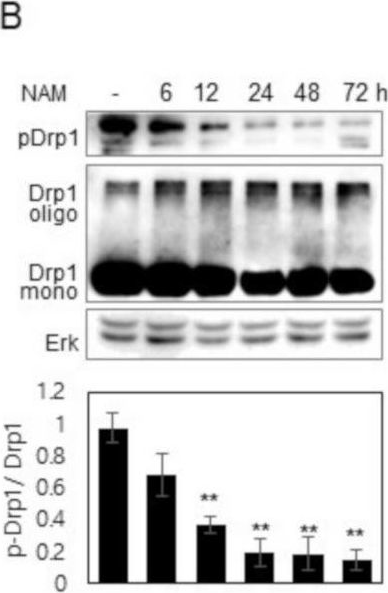
Fig.1.B
-
WB
-
Collected and cropped from Cells by CiteAb, provided under a CC-BY license
Image 1 of 53
In Cells on 10 March 2021 by Song, S. B., Park, J. S., et al.
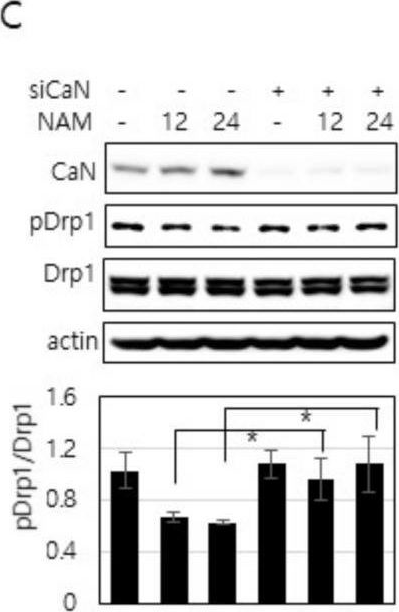
Fig.6.C
-
WB
-
Collected and cropped from Cells by CiteAb, provided under a CC-BY license
Image 1 of 53
In J Cachexia Sarcopenia Muscle on 1 June 2020 by Shah, D. S., Nisr, R. B., et al.
Fig.7.C
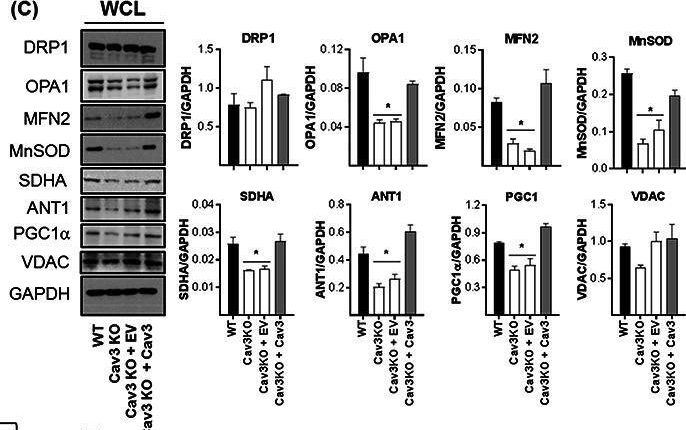
-
WB
-
Collected and cropped from Journal of Cachexia, Sarcopenia and Muscle by CiteAb, provided under a CC-BY license
Image 1 of 53
In J Cachexia Sarcopenia Muscle on 1 June 2020 by Shah, D. S., Nisr, R. B., et al.
Fig.4.A
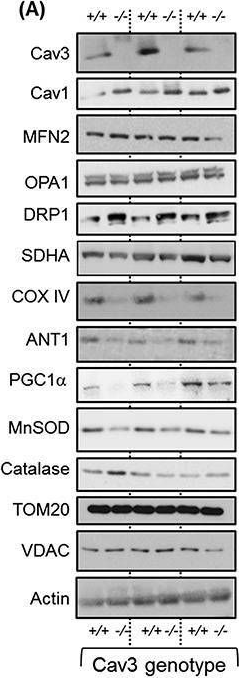
-
WB
-
Collected and cropped from Journal of Cachexia, Sarcopenia and Muscle by CiteAb, provided under a CC-BY license
Image 1 of 53
In Cell Mol Life Sci on 1 December 2019 by Nisr, R. B., Shah, D. S., et al.
Fig.8.A
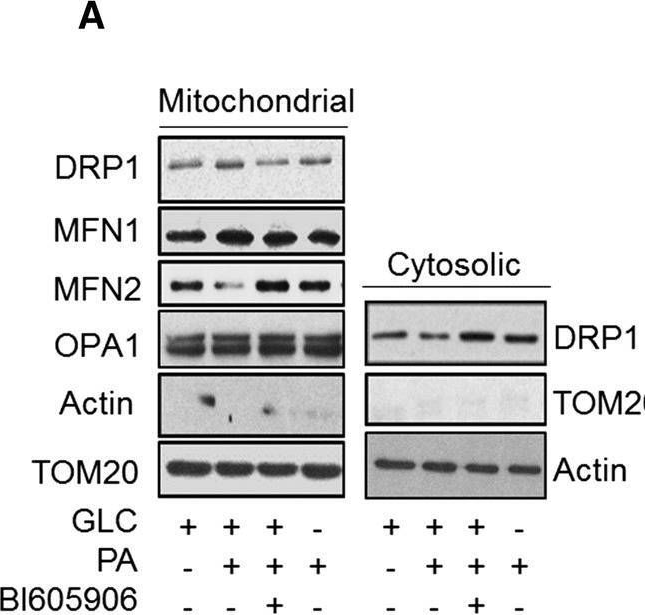
-
WB
-
Collected and cropped from Cellular and Molecular Life Sciences : CMLS by CiteAb, provided under a CC-BY license
Image 1 of 53
In J Clin Med on 21 October 2019 by Ko, H. J., Chiou, S. J., et al.
Fig.2.B
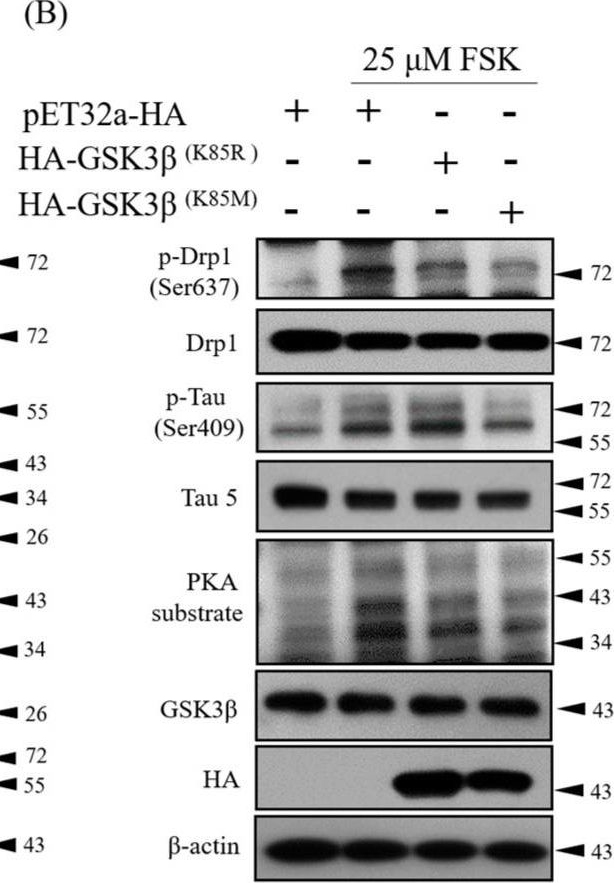
-
WB
-
Homo sapiens (Human)
Collected and cropped from Journal of Clinical Medicine by CiteAb, provided under a CC-BY license
Image 1 of 53
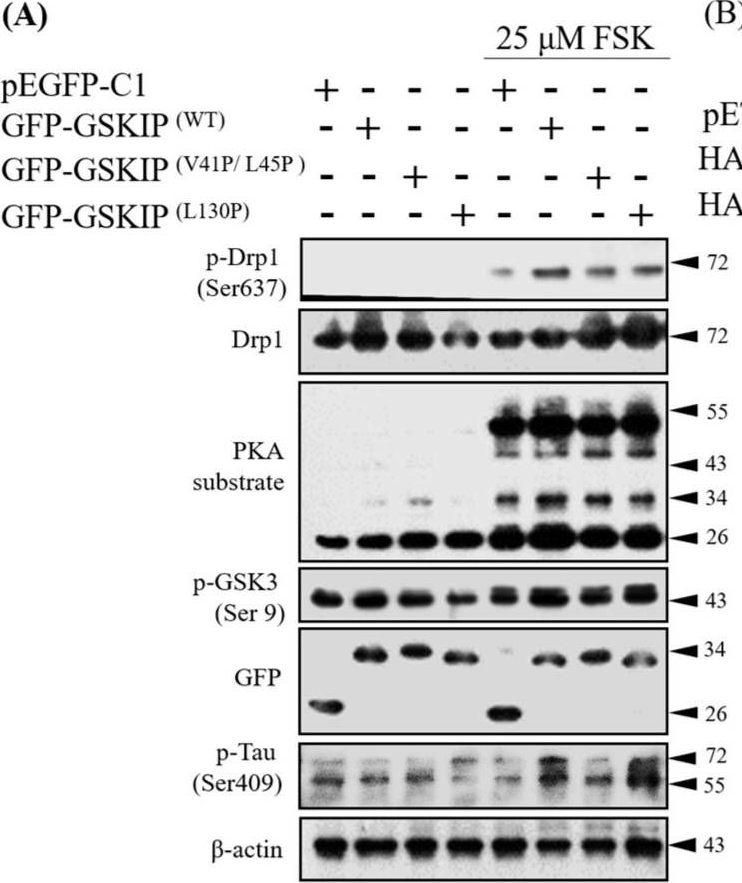
In J Clin Med on 21 October 2019 by Ko, H. J., Chiou, S. J., et al.
Fig.2.A
-
WB
-
Homo sapiens (Human)
Collected and cropped from Journal of Clinical Medicine by CiteAb, provided under a CC-BY license
Image 1 of 53
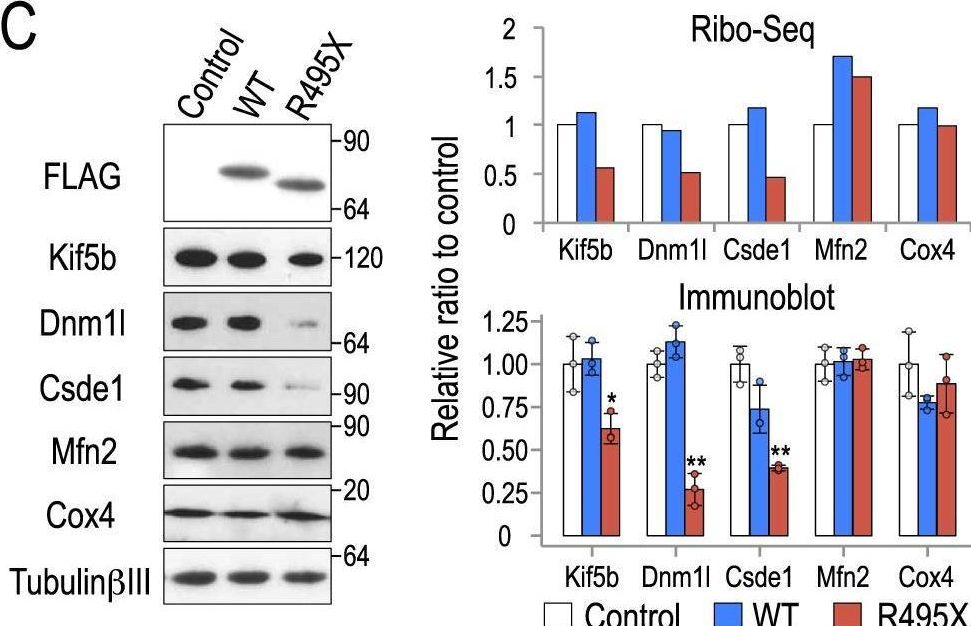
In Sci Rep on 22 October 2018 by Nakaya, T. & Maragkakis, M.
Fig.4.C
-
WB
-
Collected and cropped from Scientific Reports by CiteAb, provided under a CC-BY license
Image 1 of 53
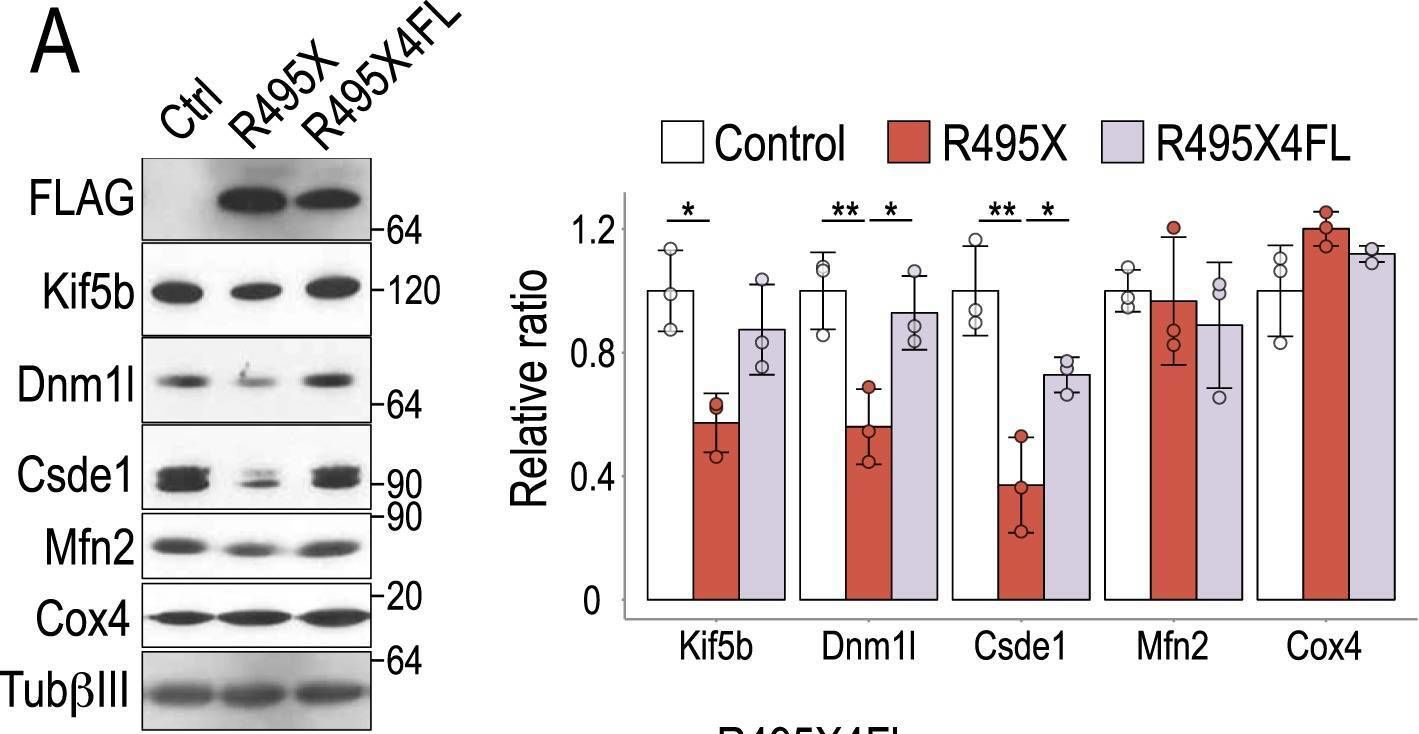
In Sci Rep on 22 October 2018 by Nakaya, T. & Maragkakis, M.
Fig.7.A
-
WB
-
Collected and cropped from Scientific Reports by CiteAb, provided under a CC-BY license
Image 1 of 53
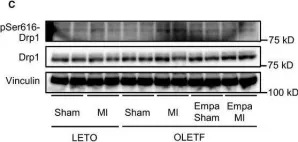
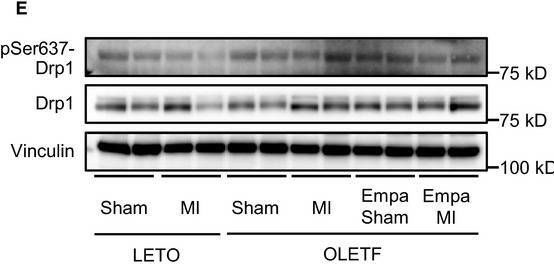
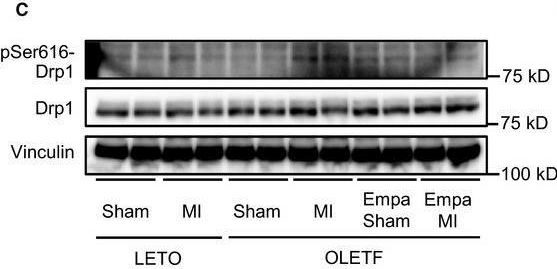
In Physiol Rep on 1 June 2018 by Mizuno, M., Kuno, A., et al.
Fig.5.E
-
WB
-
Collected and cropped from Physiological Reports by CiteAb, provided under a CC-BY license
Image 1 of 53
In Physiol Rep on 1 June 2018 by Mizuno, M., Kuno, A., et al.
Fig.5.C
-
WB
-
Collected and cropped from Physiological Reports by CiteAb, provided under a CC-BY license
Image 1 of 53
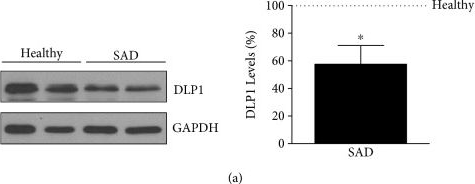
In Oxid Med Cell Longev on 5 December 2017 by Martín-Maestro, P., Gargini, R., et al.
Fig.2.A
-
WB
-
Mus musculus (House mouse)
Collected and cropped from Oxidative Medicine and Cellular Longevity by CiteAb, provided under a CC-BY license
Image 1 of 53
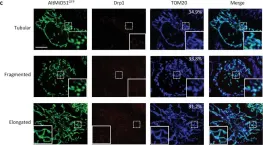
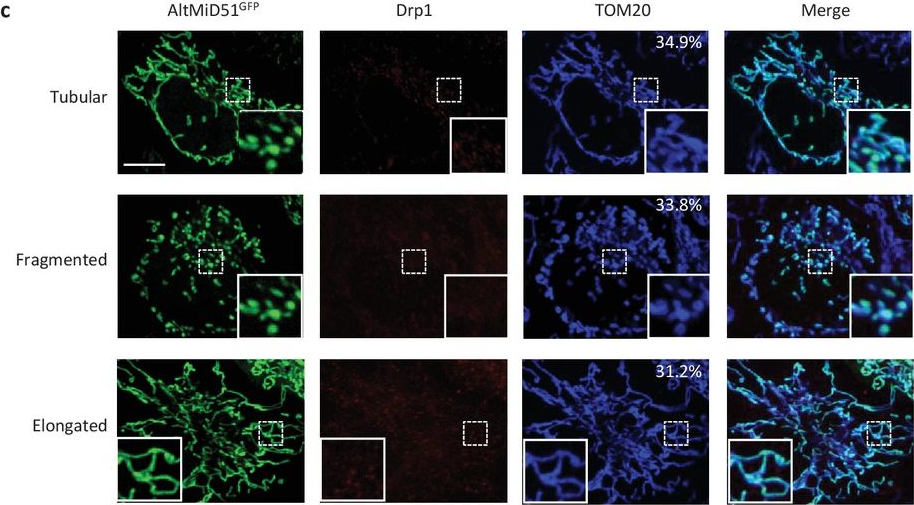
In Elife on 30 October 2017 by Samandi, S., Roy, A. V., et al.
Fig.13.C
-
ICC-IF
-
Collected and cropped from eLife by CiteAb, provided under a CC-BY license
Image 1 of 53