
Alzheimer's disease has a higher incidence in older women, with a spike in cognitive decline that tracks with visceral adiposity, dysregulated energy homeostasis and bone loss during the menopausal transition1,2. Inhibiting the action of follicle-stimulating hormone (FSH) reduces body fat, enhances thermogenesis, increases bone mass and lowers serum cholesterol in mice3-7. Here we show that FSH acts directly on hippocampal and cortical neurons to accelerate amyloid-β and Tau deposition and impair cognition in mice displaying features of Alzheimer's disease. Blocking FSH action in these mice abrogates the Alzheimer's disease-like phenotype by inhibiting the neuronal C/EBPβ-δ-secretase pathway. These data not only suggest a causal role for rising serum FSH levels in the exaggerated Alzheimer's disease pathophysiology during menopause, but also reveal an opportunity for treating Alzheimer's disease, obesity, osteoporosis and dyslipidaemia with a single FSH-blocking agent.
© 2022. The Author(s), under exclusive licence to Springer Nature Limited.
Product Citations: 12
FSH blockade improves cognition in mice with Alzheimer's disease.
In Nature on 1 March 2022 by Xiong, J., Kang, S. S., et al.
-
Neuroscience
In Nature Communications on 23 November 2021 by Cheng, H., Sebaa, R., et al.
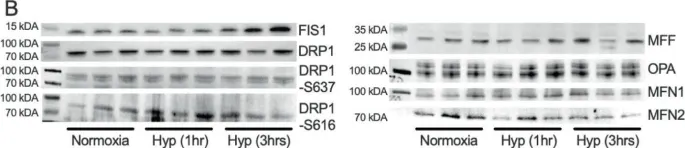
Naked mole-rats are among the most hypoxia-tolerant mammals. During hypoxia, their body temperature (Tb) decreases via unknown mechanisms to conserve energy. In small mammals, non-shivering thermogenesis in brown adipose tissue (BAT) is critical to Tb regulation; therefore, we hypothesize that hypoxia decreases naked mole-rat BAT thermogenesis. To test this, we measure changes in Tb during normoxia and hypoxia (7% O2; 1-3 h). We report that interscapular thermogenesis is high in normoxia but ceases during hypoxia, and Tb decreases. Furthermore, in BAT from animals treated in hypoxia, UCP1 and mitochondrial complexes I-V protein expression rapidly decrease, while mitochondria undergo fission, and apoptosis and mitophagy are inhibited. Finally, UCP1 expression decreases in hypoxia in three other social African mole-rat species, but not a solitary species. These findings suggest that the ability to rapidly down-regulate thermogenesis to conserve oxygen in hypoxia may have evolved preferentially in social species.
© 2021. The Author(s).
-
WB
-
Rattus norvegicus (Rat)
In BMC Biology on 25 August 2021 by Jia, T., Jacquet, T., et al.
Angiogenesis is the process by which new blood vessels arise from pre-existing ones. Fibroblast growth factor-2 (FGF-2), a leading member of the FGF family of heparin-binding growth factors, contributes to normal as well as pathological angiogenesis. Pre-mRNA alternative splicing plays a key role in the regulation of cellular and tissular homeostasis and is highly controlled by splicing factors, including SRSFs. SRSFs belong to the SR protein family and are regulated by serine/threonine kinases such as SRPK1. Up to now, the role of SR proteins and their regulators in the biology of endothelial cells remains elusive, in particular upstream signals that control their expression.
By combining 2D endothelial cells cultures, 3D collagen sprouting assay, a model of angiogenesis in cellulose sponges in mice and a model of angiogenesis in zebrafish, we collectively show that FGF-2 promotes proliferation, survival, and sprouting of endothelial cells by activating a SRSF1/SRSF3/SRPK1-dependent axis. In vitro, we further demonstrate that this FGF-2-dependent signaling pathway controls VEGFR1 pre-mRNA splicing and leads to the generation of soluble VEGFR1 splice variants, in particular a sVEGFR1-ex12 which retains an alternative last exon, that contribute to FGF-2-mediated angiogenic functions. Finally, we show that sVEGFR1-ex12 mRNA level correlates with that of FGF-2/FGFR1 in squamous lung carcinoma patients and that sVEGFR1-ex12 is a poor prognosis marker in these patients.
We demonstrate that FGF-2 promotes angiogenesis by activating a SRSF1/SRSF3/SRPK1 network that regulates VEGFR1 alternative splicing in endothelial cells, a process that could also contribute to lung tumor progression.
© 2021. The Author(s).
-
WB
In Hepatology on 1 July 2021 by Khatun, M., Sur, S., et al.
HCV often causes chronic infection in liver, cirrhosis, and, in some instances, HCC. HCV encodes several factors' those impair host genes for establishment of chronic infection. The long noncoding RNAs (lncRNAs) display diverse effects on biological regulations. However, their role in virus replication and underlying diseases is poorly understood. In this study, we have shown that HCV exploits lncRNA long intergenic nonprotein-coding RNA, p53 induced transcript (Linc-Pint) in hepatocytes for enhancement of lipogenesis.
We identified a lncRNA, Linc-Pint, which is significantly down-regulated in HCV-replicating hepatocytes and liver specimens from HCV infected patients. Using RNA pull-down proteomics, we identified serine/arginine protein specific kinase 2 (SRPK2) as an interacting partner of Linc-Pint. A subsequent study demonstrated that overexpression of Linc-Pint inhibits the expression of lipogenesis-related genes, such as fatty acid synthase and ATP-citrate lyase. We also observed that Linc-Pint significantly inhibits HCV replication. Furthermore, HCV-mediated enhanced lipogenesis can be controlled by exogenous Linc-Pint expression. Together, our results suggested that HCV-mediated down-regulation of Linc-Pint enhances lipogenesis favoring virus replication and liver disease progression.
We have shown that SRPK2 is a direct target of Linc-Pint and that depletion of SRPK2 inhibits lipogenesis. Our study contributes to the mechanistic understanding of the role of Linc-Pint in HCV-associated liver pathogenesis.
© 2020 by the American Association for the Study of Liver Diseases.
-
WB
-
Homo sapiens (Human)
-
Genetics
-
Immunology and Microbiology
In Developmental Cell on 7 December 2020 by Bustos, F., Segarra-Fas, A., et al.
Conserved protein kinases with core cellular functions have been frequently redeployed during metazoan evolution to regulate specialized developmental processes. The Ser/Arg (SR)-rich splicing factor (SRSF) protein kinase (SRPK), which is implicated in splicing regulation, is one such conserved eukaryotic kinase. Surprisingly, we show that SRPK has acquired the capacity to control a neurodevelopmental ubiquitin signaling pathway. In mammalian embryonic stem cells and cultured neurons, SRPK phosphorylates Ser-Arg motifs in RNF12/RLIM, a key developmental E3 ubiquitin ligase that is mutated in an intellectual disability syndrome. Processive phosphorylation by SRPK stimulates RNF12-dependent ubiquitylation of nuclear transcription factor substrates, thereby acting to restrain a neural gene expression program that is aberrantly expressed in intellectual disability. SRPK family genes are also mutated in intellectual disability disorders, and patient-derived SRPK point mutations impair RNF12 phosphorylation. Our data reveal unappreciated functional diversification of SRPK to regulate ubiquitin signaling that ensures correct regulation of neurodevelopmental gene expression.
Copyright © 2020 The Authors. Published by Elsevier Inc. All rights reserved.
-
Stem Cells and Developmental Biology
In Nat Commun on 23 November 2021 by Cheng, H., Sebaa, R., et al.
Fig.7.B

-
WB
-
Collected and cropped from Nat Commun by CiteAb, provided under a CC-BY license
Image 1 of 5
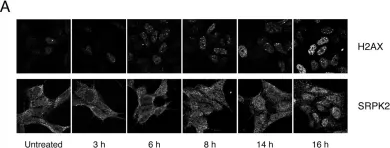
In PLoS One on 26 April 2016 by Seo, J., Singh, N. N., et al.
Fig.6.A

-
WB
-
Collected and cropped from PLoS One by CiteAb, provided under a CC-BY license
Image 1 of 5
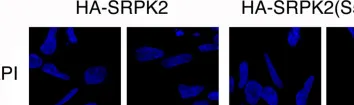
In PLoS One on 25 April 2013 by Vivarelli, S., Lenzken, S. C., et al.
Fig.5.A

-
ICC-IF
-
Homo sapiens (Human)
Collected and cropped from PLoS One by CiteAb, provided under a CC-BY license
Image 1 of 5
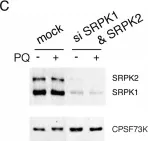
In PLoS One on 25 April 2013 by Vivarelli, S., Lenzken, S. C., et al.
Fig.3.A

-
WB
-
Homo sapiens (Human)
Collected and cropped from PLoS One by CiteAb, provided under a CC-BY license
Image 1 of 5
In PLoS One on 25 April 2013 by Vivarelli, S., Lenzken, S. C., et al.
Fig.2.C

-
WB
-
Homo sapiens (Human)
Collected and cropped from PLoS One by CiteAb, provided under a CC-BY license
Image 1 of 5