
The GARP complex is an evolutionarily conserved protein complex proposed to tether endosome-derived vesicles at the trans-Golgi network. While complete depletion of the GARP leads to severe trafficking and glycosylation defects, the primary defects linked to GARP dysfunction remain unclear. In this study, we utilized the mAID degron strategy to achieve rapid degradation of VPS54 in human cells, acutely disrupting GARP function. This resulted in the partial mislocalization and degradation of a subset of Golgi-resident proteins, including TGN46, ATP7A, TMEM87A, CPD, C1GALT1 and GS15. Enzyme recycling defects led to O-glycosylation abnormalities. Additionally, while fibronectin and cathepsin D secretion were altered, mannose-6-phosphate receptors were largely unaffected. Partial displacement of COPI, AP1 and GGA coats caused a significant accumulation of vesicle-like structures and large vacuoles. Electron microscopy detection of GARP-dependent vesicles and identifying specific cargo proteins provide direct experimental evidence of GARP's role as a vesicular tether. We conclude that the primary defects of GARP dysfunction involve vesicular coat mislocalization, accumulation of GARP-dependent vesicles, degradation and mislocalization of specific Golgi proteins and O-glycosylation defects.
© 2025 The Author(s). Traffic published by John Wiley & Sons Ltd.
Product Citations: 28
In Traffic (Copenhagen, Denmark) on 18 March 2025 by Khakurel, A., Pokrovskaya, I., et al.
-
Cell Biology
Alternative autophagy dampens UVB-induced NLRP3 inflammasome activation in human keratinocytes.
In The Journal of Biological Chemistry on 1 April 2024 by Hasegawa, T., Noguchi, S., et al.
Sunlight exposure results in an inflammatory reaction of the skin commonly known as sunburn, which increases skin cancer risk. In particular, the ultraviolet B (UVB) component of sunlight induces inflammasome activation in keratinocytes to instigate the cutaneous inflammatory responses. Here, we explore the intracellular machinery that maintains skin homeostasis by suppressing UVB-induced inflammasome activation in human keratinocytes. We found that pharmacological inhibition of autophagy promoted UVB-induced NLRP3 inflammasome activation. Unexpectedly, however, gene silencing of Atg5 or Atg7, which are critical for conventional autophagy, had no effect, whereas gene silencing of Beclin1, which is essential not only for conventional autophagy but also for Atg5/Atg7-independent alternative autophagy, promoted UVB-induced inflammasome activation, indicating an involvement of alternative autophagy. We found that damaged mitochondria were highly accumulated in UVB-irradiated keratinocytes when alternative autophagy was inhibited, and they appear to be recognized by NLRP3. Overall, our findings indicate that alternative autophagy, rather than conventional autophagy, suppresses UVB-induced NLRP3 inflammasome activation through the clearance of damaged mitochondria in human keratinocytes and illustrate a previously unknown involvement of alternative autophagy in inflammation. Alternative autophagy may be a new therapeutic target for sunburn and associated cutaneous disorders.
Copyright © 2024 The Authors. Published by Elsevier Inc. All rights reserved.
-
ICC
-
Homo sapiens (Human)
-
Biochemistry and Molecular biology
-
Cell Biology
Phosphatidylserine synthesis controls oncogenic B cell receptor signaling in B cell lymphoma.
In The Journal of Cell Biology on 5 February 2024 by Omi, J., Kato, T., et al.
Cancer cells harness lipid metabolism to promote their own survival. We screened 47 cancer cell lines for survival dependency on phosphatidylserine (PS) synthesis using a PS synthase 1 (PTDSS1) inhibitor and found that B cell lymphoma is highly dependent on PS. Inhibition of PTDSS1 in B cell lymphoma cells caused a reduction of PS and phosphatidylethanolamine levels and an increase of phosphoinositide levels. The resulting imbalance of the membrane phospholipidome lowered the activation threshold for B cell receptor (BCR), a B cell-specific survival mechanism. BCR hyperactivation led to aberrant elevation of downstream Ca2+ signaling and subsequent apoptotic cell death. In a mouse xenograft model, PTDSS1 inhibition efficiently suppressed tumor growth and prolonged survival. Our findings suggest that PS synthesis may be a critical vulnerability of malignant B cell lymphomas that can be targeted pharmacologically.
© 2023 Omi et al.
-
Cancer Research
-
Cell Biology
-
Immunology and Microbiology
Syntaxin-5's flexibility in SNARE pairing supports Golgi functions.
In Traffic (Copenhagen, Denmark) on 1 August 2023 by D'Souza, Z., Pokrovskaya, I., et al.
Deficiency in the conserved oligomeric Golgi (COG) complex that orchestrates SNARE-mediated tethering/fusion of vesicles that recycle the Golgi's glycosylation machinery results in severe glycosylation defects. Although two major Golgi v-SNAREs, GS28/GOSR1, and GS15/BET1L, are depleted in COG-deficient cells, the complete knockout of GS28 and GS15 only modestly affects Golgi glycosylation, indicating the existence of an adaptation mechanism in Golgi SNARE. Indeed, quantitative mass-spectrometry analysis of STX5-interacting proteins revealed two novel Golgi SNARE complexes-STX5/SNAP29/VAMP7 and STX5/VTI1B/STX8/YKT6. These complexes are present in wild-type cells, but their usage is significantly increased in both GS28- and COG-deficient cells. Upon GS28 deletion, SNAP29 increased its Golgi residency in a STX5-dependent manner. While STX5 depletion and Retro2-induced diversion from the Golgi severely affect protein glycosylation, GS28/SNAP29 and GS28/VTI1B double knockouts alter glycosylation similarly to GS28 KO, indicating that a single STX5-based SNARE complex is sufficient to support Golgi glycosylation. Importantly, co-depletion of three Golgi SNARE complexes in GS28/SNAP29/VTI1B TKO cells resulted in severe glycosylation defects and a reduced capacity for glycosylation enzyme retention at the Golgi. This study demonstrates the remarkable plasticity in SXT5-mediated membrane trafficking, uncovering a novel adaptive response to the failure of canonical intra-Golgi vesicle tethering/fusion machinery.
© 2023 The Authors. Traffic published by John Wiley & Sons Ltd.
-
Cell Biology
In Frontiers in Cell and Developmental Biology on 30 December 2022 by Khakurel, A., Kudlyk, T., et al.
Golgi-associated retrograde protein (GARP) is an evolutionary conserved heterotetrameric protein complex that tethers endosome-derived vesicles and is vital for Golgi glycosylation. Microscopy and proteomic approaches were employed to investigate defects in Golgi physiology in RPE1 cells depleted for the GARP complex. Both cis and trans-Golgi compartments were significantly enlarged in GARP-knock-out (KO) cells. Proteomic analysis of Golgi-enriched membranes revealed significant depletion of a subset of Golgi residents, including Ca2+ binding proteins, enzymes, and SNAREs. Validation of proteomics studies revealed that SDF4 and ATP2C1, related to Golgi calcium homeostasis, as well as intra-Golgi v-SNAREs GOSR1 and BET1L, were significantly depleted in GARP-KO cells. Finding that GARP-KO is more deleterious to Golgi physiology than deletion of GARP-sensitive v-SNAREs, prompted a detailed investigation of COPI trafficking machinery. We discovered that in GARP-KO cells COPI is significantly displaced from the Golgi and partially relocalized to the ER-Golgi intermediate compartment (ERGIC). Moreover, COPI accessory proteins GOLPH3, ARFGAP1, GBF1, and BIG1 are also relocated to off-Golgi compartments. We propose that the dysregulation of COPI machinery, along with the depletion of Golgi v-SNAREs and alteration of Golgi Ca2+ homeostasis, are the major driving factors for the depletion of Golgi resident proteins, structural alterations, and glycosylation defects in GARP deficient cells.
Copyright © 2022 Khakurel, Kudlyk, Pokrovskaya, D’Souza and Lupashin.
-
Cell Biology
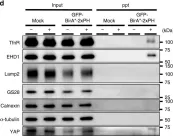
In JCI Insight on 8 February 2021 by Hu, W., Liu, Z., et al.
Fig.5.A

-
WB
-
Collected and cropped from JCI Insight by CiteAb, provided under a CC-BY license
Image 1 of 5
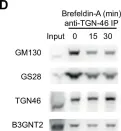
In Nat Commun on 1 November 2017 by Matsudaira, T., Mukai, K., et al.
Fig.1.D

-
WB
-
Collected and cropped from Nat Commun by CiteAb, provided under a CC-BY license
Image 1 of 5
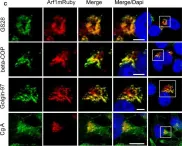
In Elife on 8 June 2016 by Mkhikian, H., Mortales, C. L., et al.
Fig.6.D

-
WB
-
Homo sapiens (Human)
Collected and cropped from Elife by CiteAb, provided under a CC-BY license
Image 1 of 5
In J Cell Mol Med on 1 May 2015 by Münzberg, C., Höhn, K., et al.
Fig.1.C

-
ICC-IF
-
Homo sapiens (Human)
Collected and cropped from J Cell Mol Med by CiteAb, provided under a CC-BY license
Image 1 of 5
In BMC Genomics on 27 December 2007 by Schulz, T. C., Swistowska, A. M., et al.
Fig.4.D

-
ICC-IF
-
Homo sapiens (Human)
Collected and cropped from BMC Genomics by CiteAb, provided under a CC-BY license
Image 1 of 5