Genetic hypomorphic defects in X chromosomal IKBKG coding for the NF-κB essential modulator (NEMO) lead to ectodermal dysplasia and immunodeficiency in males and the skin disorder incontinentia pigmenti (IP) in females, respectively. NF-κB essential modulator (NEMO) Δ-exon 5-autoinflammatory syndrome (NEMO-NDAS) is a systemic autoinflammatory disease caused by alternative splicing and increased proportion of NEMO-Δex5. We investigated a female carrier presenting with IP and NEMO-NDAS due to non-skewed X-inactivation.
IKBKG transcripts were quantified in peripheral blood mononuclear cells isolated from the patient, her mother, and healthy controls using RT-PCR and nanopore sequencing. Corresponding proteins were analyzed by western blotting and flow cytometry. Besides toll-like receptor (TLR) and tumor necrosis factor (TNF) signaling, the interferon signature, cytokine production and X-inactivation status were investigated.
IP and autoinflammation with recurrent fever, oral ulcers, hepatitis, and neutropenia, but no immunodeficiency was observed in a female patient. Besides moderately reduced NEMO signaling function, type I interferonopathy, and elevated IL-18 and CXCL10 were found. She and her mother both carried the heterozygous variant c.613 C > T p.(Gln205*) in exon 5 of IKBKG previously reported in NEMO-deficient patients. However, X-inactivation was skewed in the mother, but not in the patient. Alternative splicing led to increased ratios of NEMO-Dex5 over full-length protein in peripheral blood cell subsets causing autoinflammation. Clinical symptoms partially resolved under treatment with TNF inhibitors.
Non-skewed X-inactivation can lead to NEMO-NDAS in females with IP carrying hypomorphic IKBKG variants due to alternative splicing and increased proportions of NEMO-∆ex5.
© 2024. The Author(s).
Product Citations: 26
In Journal of Clinical Immunology on 12 September 2024 by Eigemann, J., Janda, A., et al.
-
WB
-
Homo sapiens (Human)
Damaged mitochondria recruit the effector NEMO to activate NF-κB signaling.
In Molecular Cell on 7 September 2023 by Harding, O., Holzer, E., et al.
Failure to clear damaged mitochondria via mitophagy disrupts physiological function and may initiate damage signaling via inflammatory cascades, although how these pathways intersect remains unclear. We discovered that nuclear factor kappa B (NF-κB) essential regulator NF-κB effector molecule (NEMO) is recruited to damaged mitochondria in a Parkin-dependent manner in a time course similar to recruitment of the structurally related mitophagy adaptor, optineurin (OPTN). Upon recruitment, NEMO partitions into phase-separated condensates distinct from OPTN but colocalizing with p62/SQSTM1. NEMO recruitment, in turn, recruits the active catalytic inhibitor of kappa B kinase (IKK) component phospho-IKKβ, initiating NF-κB signaling and the upregulation of inflammatory cytokines. Consistent with a potential neuroinflammatory role, NEMO is recruited to mitochondria in primary astrocytes upon oxidative stress. These findings suggest that damaged, ubiquitinated mitochondria serve as an intracellular platform to initiate innate immune signaling, promoting the formation of activated IKK complexes sufficient to activate NF-κB signaling. We propose that mitophagy and NF-κB signaling are initiated as parallel pathways in response to mitochondrial stress.
Copyright © 2023 The Authors. Published by Elsevier Inc. All rights reserved.
-
Homo sapiens (Human)
-
Biochemistry and Molecular biology
-
Cell Biology
TRAF6 initiates inflammatory signaling via organizing membraneless cytoplasmic condensates
Preprint on BioRxiv : the Preprint Server for Biology on 23 June 2023 by Wang, J., Wang, J., et al.
The tumor necrosis factor receptor-associated factor 6 (TRAF6) is a central molecule in multiple signaling pathways, i.e. the TNF receptor, the Toll-like receptors (TLRs), and interleukin-1 receptor (IL-1R) pathways. Upon pathogen associated molecular patterns (PAMPs) or damage associated molecular patterns (DAMPs) stimulations, TRAF6 activates downstream of NF-κB signaling. However, the precise mechanism of how TRAF6 activates downstream molecules remains unclear. Here, we demonstrate that TRAF6 acts as a sensor for upstream signals to initiate liquid-liquid phase separation (LLPS) of itself and downstream proteins, forming membraneless condensates. Subsequent recruitment, enrichment and activation of downstream effectors in the condensates lead to robust inflammatory signal transduction. The multivalent interactions mediated by its RING domain, zinc finger domain 1, and coiled-coil domain mediates the LLPS process. Forced phase separation of TRAF6 induced NF-κB activation. Disruption of TRAF6 phase separation abrogates activation of NF-κB signaling. Overall, we uncover the spatial organization of molecules by TRAF6 through phase separation as a subcellular platform to activate inflammatory signaling. Targeting TRAF6 phase separation hold promises for therapeutics aiming autoimmune diseases, inflammation and cancers.
-
Cell Biology
-
Immunology and Microbiology
In Science Advances on 22 March 2023 by Riera-Domingo, C., Leite-Gomes, E., et al.
Endothelial cells (ECs) grant access of disseminated cancer cells to distant organs. However, the molecular players regulating the activation of quiescent ECs at the premetastatic niche (PMN) remain elusive. Here, we find that ECs at the PMN coexpress tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) and its cognate death receptor 5 (DR5). Unexpectedly, endothelial TRAIL interacts intracellularly with DR5 to prevent its signaling and preserve a quiescent vascular phenotype. In absence of endothelial TRAIL, DR5 activation induces EC death and nuclear factor κB/p38-dependent EC stickiness, compromising vascular integrity and promoting myeloid cell infiltration, breast cancer cell adhesion, and metastasis. Consistently, both down-regulation of endothelial TRAIL at the PMN by proangiogenic tumor-secreted factors and the presence of the endogenous TRAIL inhibitors decoy receptor 1 (DcR1) and DcR2 favor metastasis. This study discloses an intracrine mechanism whereby TRAIL blocks DR5 signaling in quiescent endothelia, acting as gatekeeper of the vascular barrier that is corrupted by the tumor during cancer cell dissemination.
-
WB
-
Mus musculus (House mouse)
-
Cancer Research
In IScience on 21 October 2022 by Laplantine, E., Chable-Bessia, C., et al.
Patients with severe COVID-19 show an altered immune response that fails to control the viral spread and suffer from exacerbated inflammatory response, which eventually can lead to death. A major challenge is to develop an effective treatment for COVID-19. NF-κB is a major player in innate immunity and inflammatory process. By a high-throughput screening approach, we identified FDA-approved compounds that inhibit the NF-κB pathway and thus dampen inflammation. Among these, we show that Auranofin prevents post-translational modifications of NF-κB effectors and their recruitment into activating complexes in response to SARS-CoV-2 infection or cytokine stimulation. In addition, we demonstrate that Auranofin counteracts several steps of SARS-CoV-2 infection. First, it inhibits a raft-dependent endocytic pathway involved in SARS-CoV-2 entry into host cells; Second, Auranofin alters the ACE2 mobility at the plasma membrane. Overall, Auranofin should prevent SARS-CoV-2 infection and inflammatory damages, offering new opportunities as a repurposable drug candidate to treat COVID-19.
© 2022 The Authors.
-
COVID-19
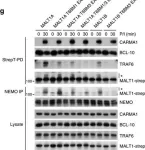
In Nat Commun on 12 April 2016 by Meininger, I., Griesbach, R. A., et al.
Fig.1.G

-
WB
-
Collected and cropped from Nat Commun by CiteAb, provided under a CC-BY license
Image 1 of 1