DNA double-strand breaks (DSBs) are toxic lesions that can lead to genome instability if not properly repaired. Breaks incurred in G1 phase of the cell cycle are predominantly fixed by non-homologous end-joining (NHEJ), while homologous recombination (HR) is the primary repair pathway in S and G2. Microhomology-mediated end-joining (MMEJ) is intrinsically error-prone and considered a backup DSB repair pathway that becomes essential when HR and NHEJ are compromised. In this study, we uncover MMEJ as the major DSB repair pathway in M phase. Using CRISPR/Cas9-based synthetic lethal screens, we identify subunits of the 9-1-1 complex (RAD9A-HUS1-RAD1) and its interacting partner, RHINO, as critical MMEJ factors. Mechanistically, we show that the function of 9-1-1 and RHINO in MMEJ is inconsistent with their well-established role in ATR signaling. Instead, RHINO plays an unexpected and essential role in directing mutagenic repair to M phase by directly binding to Polymerase theta (Polθ) and promoting its recruitment to DSBs in mitosis. In addition, we provide evidence that mitotic MMEJ repairs persistent DNA damage that originates in S phase but is not repaired by HR. The latter findings could explain the synthetic lethal relationship between POLQ and BRCA1/2 and the synergistic effect of Polθ and PARP inhibitors. In summary, our study identifies MMEJ as the primary pathway for repairing DSBs during mitosis and highlights an unanticipated role for RHINO in directing mutagenic repair to M phase.
Product Citations: 9
RHINO restricts MMEJ activity to mitosis
Preprint on BioRxiv : the Preprint Server for Biology on 16 March 2023 by Brambati, A., Sacco, O., et al.
-
WB
-
Cell Biology
In Cellular Signalling on 1 October 2021 by Panigrahi, S. K., Broustas, C. G., et al.
Metastatic progression is the key feature of prostate cancer primarily responsible for mortality caused by this disease. RAD9 is an oncogene for prostate cancer, and the encoded protein enhances metastasis-related phenotypes. RAD9 is a transcription factor with a limited set of regulated target genes, but the complete list of downstream genes critical for prostate carcinogenesis is unknown. We used microarray gene expression profiling and chromatin immunoprecipitation in parallel to identify genes transcriptionally controlled by RAD9 that contribute to this cancer. We found expression of 44 genes altered in human prostate cancer DU145 cells when RAD9 is knocked down by siRNA, and all of them bind RAD9 at their genomic location. FOXP1 and NDRG1 were down regulated when RAD9 expression was reduced, and we evaluated them further. We demonstrate that reduced RAD9, FOXP1 or NDGR1 expression decreases cell proliferation, rapid migration, anchorage-independent growth, anoikis resistance, and aerobic glycolysis. Ectopic expression of FOXP1 or NDRG1 partially restored aerobic glycolysis to prostate cancer cells with reduced RAD9 abundance, but only FOXP1 significantly complemented the other deficiencies. We thus show, for the first time, that RAD9 regulates FOXP1 and NDRG1 expression, and they function differently as downstream effectors for RAD9-mediated prostate cancer cell activities.
Copyright © 2021 Elsevier Inc. All rights reserved.
-
WB
-
Cancer Research
-
Cell Biology
In Carcinogenesis on 25 February 2021 by Zhu, A., Hopkins, K. M., et al.
Prostate cancer is the second most common type of cancer and the second leading cause of cancer death in American men. RAD9 stabilizes the genome, but prostate cancer cells and tumors often have high quantities of the protein. Reduction of RAD9 level within prostate cancer cells decreases tumorigenicity of nude mouse xenographs and metastasis phenotypes in culture, indicating that RAD9 overproduction is essential for the disease. In prostate cancer DU145 cells, CpG hypermethylation in a transcription suppressor site of RAD9 intron 2 causes high-level gene expression. Herein, we demonstrate that DNA methyltransferases DNMT1 and DNMT3B are highly abundant in prostate cancer cells DU145, CWR22, LNCaP and PC-3; yet, these DNMTs bind primarily to the transcription suppressor in DU145, the only cells where methylation is critical for RAD9 regulation. For DU145 cells, DNMT1 or DNMT3B shRNA reduced RAD9 level and tumorigenicity, and RAD9 ectopic expression restored this latter activity in the DNMT knockdown cells. High levels of RAD9, DNMT1, DNMT3B and RAD9 transcription suppressor hypermethylation were significantly correlated in prostate tumors, and not in normal prostate tissues. Based on these results, we propose a novel model where RAD9 is regulated epigenetically by DNMT1 and DNMT3B, via targeted hypermethylation, and that consequent RAD9 overproduction promotes prostate tumorigenesis.
© The Author(s) 2020. Published by Oxford University Press. All rights reserved. For Permissions, please email: journals.permissions@oup.com.
-
Cancer Research
In Carcinogenesis on 12 March 2019 by Broustas, C. G., Hopkins, K. M., et al.
RAD9A plays an important role in prostate tumorigenesis and metastasis-related phenotypes. The protein classically functions as part of the RAD9A-HUS1-RAD1 complex but can also act independently. RAD9A can selectively transactivate multiple genes, including CDKN1A and NEIL1 by binding p53-consensus sequences in or near promoters. RAD9A is overexpressed in human prostate cancer specimens and cell lines; its expression correlates with tumor progression. Silencing RAD9A in prostate cancer cells impairs their ability to form tumors in vivo and migrate as well as grow anchorage independently in vitro. We demonstrate herein that RAD9A transcriptionally controls AGR2, a gene aberrantly overexpressed in patients with metastatic prostate cancer. Transient or stable knockdown of RAD9A in PC-3 cells caused downregulation of AGR2 protein abundance. Reduced AGR2 protein levels were due to lower abundance of AGR2 mRNA. The AGR2 genomic region upstream of the coding initiation site contains several p53 consensus sequences. RAD9A bound specifically to the 5'-untranslated region of AGR2 in PC-3 cells at a partial p53 consensus sequence at position +3136 downstream from the transcription start site, determined by chromatin immunoprecipitation, followed by PCR amplification. Binding of RAD9A to the p53 consensus sequence was sufficient to drive AGR2 gene transcription, shown by a luciferase reporter assay. In contrast, when the RAD9A-binding sequence on the AGR2 was mutated, no luciferase activity was detected. Knockdown of RAD9A in PC-3 cells impaired cell migration and anchorage-independent growth. However, ectopically expressed AGR2 in RAD9A-depleted PC-3 cells restored these phenotypes. Our results suggest RAD9A drives metastasis by controlling AGR2 abundance.
© The Author(s) 2018. Published by Oxford University Press. All rights reserved. For Permissions, please email: journals.permissions@oup.com.
-
WB
-
Biochemistry and Molecular biology
-
Cancer Research
In PLoS ONE on 15 December 2015 by Osorio-Zambrano, W. F. & Davey, S.
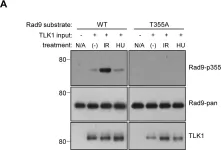
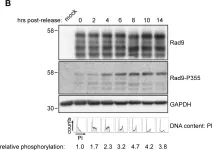
Phosphorylation of Rad9A at S387 is critical for establishing a physical interaction with TopBP1, and to downstream activation of Chk1 for checkpoint activation. We have previously demonstrated a phosphorylation of Rad9A that occurs at late time points in cells exposed to genotoxic agents, which is eliminated by either Rad9A overexpression, or conversion of S387 to a non-phosphorylatable analogue. Based on this, we hypothesized that this late Rad9A phosphorylation is part of a feedback loop regulating the checkpoint. Here, we show that Rad9A is hyperphosphorylated and accumulates in cells exposed to bleomycin. Following the removal of bleomycin, Rad9A is polyubiquitinated, and Rad9A protein levels drop, indicating an active degradation process for Rad9A. Chk1 inhibition by UCN-01 or siRNA reduces Rad9A levels in cells synchronized in S-phase or exposed to DNA damage, indicating that Chk1 activation is required for Rad9A stabilization in S-phase and during checkpoint activation. Together, these results demonstrate a positive feedback loop involving Rad9A-dependend activation of Chk1, coupled with Chk1-dependent stabilization of Rad9A that is critical for checkpoint regulation.
-
WB
-
Homo sapiens (Human)
In PLoS One on 1 January 2014 by Kelly, R. & Davey, S. K.
Fig.4.A

-
WB
-
Homo sapiens (Human)
Collected and cropped from PLoS One by CiteAb, provided under a CC-BY license
Image 1 of 2
In PLoS One on 1 January 2014 by Kelly, R. & Davey, S. K.
Fig.4.B

-
WB
-
Homo sapiens (Human)
Collected and cropped from PLoS One by CiteAb, provided under a CC-BY license
Image 1 of 2