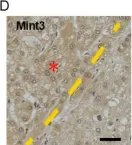
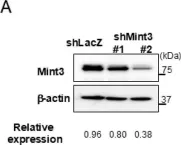
Mint3 enhances aerobic ATP production with subsequent nuclear translocation of hypoxia-inducible factor-1 (HIF-1) and activation of angiogenesis-related genes. It remains unclear if and when Mint3 is activated and whether it is involved in hepatocarcinogenesis. We explored the expression of Mint3 in surgically resected hepatocellular carcinoma (HCC) tissues. We evaluated the effects of Mint3 knockdown on spheroid formation capacity and subcutaneous tumor growth in immune-deficient mice. We used Mint3 knockout mice to evaluate the effects of chemically induced HCC development. Mint3 was overexpressed in well-differentiated HCC with the activation of HIF-1 target genes irrespective of the absence of hypervascularization. Mint3 knockdown ameliorated the expression of HIF-1 target genes in patient-derived HCC cell lines and suppressed spheroid formation. Mint3 knockdown further inhibited subcutaneous tumor formation in vivo in immune-deficient mice. Chemical HCC development induced by N-nitrosodiethylamine (DEN) or DEN/CCl4 was dramatically suppressed in Mint3 knockout mice compared to control mice. Mint3 plays a crucial role in early-stage HCC development before hypervascularization by activating HIF-1 target genes before the tumor becomes hypoxic. Mint3 is a molecular target that prevents HCC development in the early stages.
Product Citations: 8
Mint3 as a Molecular Target Activated in the Early Stage of Hepatocarcinogenesis.
In International Journal of Molecular Sciences on 8 February 2025 by Nishitani, M., Okada, H., et al.
-
IHC
-
WB
Pharmacological inhibition of Mint3 attenuates tumour growth, metastasis, and endotoxic shock.
In Communications Biology on 7 October 2021 by Sakamoto, T., Fukui, Y., et al.
Hypoxia-inducible factor-1 (HIF-1) plays essential roles in human diseases, though its central role in oxygen homoeostasis hinders the development of direct HIF-1-targeted pharmacological approaches. Here, we surveyed small-molecule compounds that efficiently inhibit the transcriptional activity of HIF-1 without affecting body homoeostasis. We focused on Mint3, which activates HIF-1 transcriptional activity in limited types of cells, such as cancer cells and macrophages, by suppressing the factor inhibiting HIF-1 (FIH-1). We identified naphthofluorescein, which inhibited the Mint3-FIH-1 interaction in vitro and suppressed Mint3-dependent HIF-1 activity and glycolysis in cancer cells and macrophages without evidence of cytotoxicity in vitro. In vivo naphthofluorescein administration suppressed tumour growth and metastasis without adverse effects, similar to the genetic depletion of Mint3. Naphthofluorescein attenuated inflammatory cytokine production and endotoxic shock in mice. Thus, Mint3 inhibitors may present a new targeted therapeutic option for cancer and inflammatory diseases by avoiding severe adverse effects.
© 2021. The Author(s).
-
WB
-
Homo sapiens (Human)
-
Mus musculus (House mouse)
-
Cancer Research
Mint3 is dispensable for pancreatic and kidney functions in mice.
In Biochemistry and Biophysics Reports on 1 December 2020 by Chung, Y., Saitoh, Y., et al.
Munc-18 interacting protein 3 (Mint3) is an activator of hypoxia-inducible factor-1 in cancer cells, macrophages, and cancer-associated fibroblasts under pathological conditions. However, exactly which cells highly express Mint3 in vivo and whether Mint3 depletion affects their physiological functions remain unclear. Here, we surveyed mouse tissues for specific expression of Mint3 by comparing Mint3 expression in wild-type and Mint3-knockout mice. Interestingly, immunohistochemical analyses revealed that Mint3 was highly expressed in islet cells of the pancreas, distal tubular epithelia of the kidney, choroid plexus ependymal cells of the cerebrum, medullary cells of the adrenal gland, and epithelial cells of the seminal gland. We also studied whether Mint3 depletion affects the physiological functions of the islets and kidneys. Mint3-knockout mice did not show any abnormalities in glucose-tolerance and urine-biochemical tests, indicating that Mint3 depletion was compensated for in these organs. Thus, loss of Mint3 might be compensated in the islets and kidneys under physiological conditions in mice.
© 2020 The Authors.
-
WB
-
Mus musculus (House mouse)
Mint3 potentiates TLR3/4- and RIG-I-induced IFN-β expression and antiviral immune responses.
In Proceedings of the National Academy of Sciences of the United States of America on 18 October 2016 by Huai, W., Song, H., et al.
Type I IFNs (IFN-α/β) play crucial roles in the elimination of invading viruses. Multiple immune cells including macrophages recognize viral infection through a variety of pattern recognition receptors, such as Toll-like receptors (TLRs) and retinoic acid-inducible gene-I (RIG-I)-like receptors, and initiate type I IFN secretion and subsequent antiviral immune responses. However, the mechanisms by which host immune cells can produce adequate amounts of type I IFNs and then eliminate viruses effectively remain to be further elucidated. In the present study, we show that munc18-1-interacting protein 3 (Mint3) expression can be markedly induced during viral infection in macrophages. Mint3 enhances TLR3/4- and RIG-I-induced IRF3 activation and IFN-β production by promoting K63-linked polyubiquitination of TNF receptor-associated factor 3 (TRAF3). Consistently, Mint3 deficiency greatly attenuated antiviral immune responses and increased viral replication. Therefore, we have identified Mint3 as a physiological positive regulator of TLR3/4 and RIG-I-induced IFN-β production and have outlined a feedback mechanism for the control of antiviral immune responses.
-
Mus musculus (House mouse)
-
Immunology and Microbiology
In The Journal of Biological Chemistry on 4 October 2013 by Caster, A. H. & Kahn, R. A.
The amyloid precursor protein (APP) is a ubiquitously expressed single-pass transmembrane protein that undergoes proteolytic processing by secretases to generate the pathogenic amyloid-β peptide, the major component in Alzheimer plaques. The traffic of APP through the cell determines its exposure to secretases and consequently the cleavages that generate the pathogenic or nonpathogenic peptide fragments. Despite the likely importance of APP traffic to Alzheimer disease, we still lack clear models for the routing and regulation of APP in cells. Like the traffic of most transmembrane proteins, the binding of adaptors to its cytoplasmic tail, which is 47 residues long and contains at least four distinct sorting motifs, regulates that of APP. We tested each of these for effects on the traffic of APP from the Golgi by mutating key residues within them and examining adaptor recruitment at the Golgi and traffic to post-Golgi site(s). We demonstrate strict specificity for recruitment of the Mint3 adaptor by APP at the Golgi, a critical role for Tyr-682 (within the YENPTY motif) in Mint3 recruitment and export of APP from the Golgi, and we identify LAMP1(+) structures as the proximal destination of APP after leaving the Golgi. Together, these data provide a detailed view of the first sorting step in its route to the cell surface and processing by secretases and further highlight the critical role played by Mint3.
-
Biochemistry and Molecular biology
-
Cell Biology
In Int J Mol Sci on 8 February 2025 by Nishitani, M., Okada, H., et al.
Fig.1.D

-
IHC
-
Collected and cropped from Int J Mol Sci by CiteAb, provided under a CC-BY license
Image 1 of 2
In Int J Mol Sci on 8 February 2025 by Nishitani, M., Okada, H., et al.
Fig.2.A

-
WB
-
Collected and cropped from Int J Mol Sci by CiteAb, provided under a CC-BY license
Image 1 of 2