Expansion mutations of triplet repeat sequences cause numerous inherited neurological diseases. In some diseases, affected individuals display somatic expansions in affected tissues which have been linked to accelerated disease onset and progression. There is currently considerable interest in developing therapies to slow somatic repeat expansions to delay or block disease onset. In vitro assays are particularly important to evaluate potential therapeutic interventions. Current assays typically use physical methods to monitor triplet repeat lengths within a population of cells. While useful, most of these assays are relatively slow (∼six weeks) and are somewhat limited in sensitivity to rare events. Here, a new assay, called TRX, is described to monitor CAG•CTG triplet repeat expansions more rapidly and with better sensitivity. TRX uses human tissue culture cells expressing two fluorescent proteins. Red fluorescent protein TagRFP658 is constitutively expressed and serves as an internal control. GFP is expressed in a CAG•CTG repeat length-dependent manner, with longer repeat lengths predicted to give higher green fluorescence intensity. Standard flow cytometry allows quantification of changes in fluorescent signal as a simple readout with <2% sensitivity. Two independently derived cell lines with 63 or 59 CAG repeats yielded similar rates of TRX activity. Cells with increased green fluorescence were observed within one to two weeks of culture, with longer times leading to additional signal. The appearance of green fluorescence was partly dependent on MutSβ, the DNA MSH2-MSH3 complex, based on siRNA knockdown of MSH3. However, physical analysis of the CAG•CTG repeat tracts by MiSeq deep sequencing or capillary electrophoresis showed limited changes in the length of the repeat tracts. We conclude that the TRX assay is a promising new tool for monitoring CAG•CTG repeat expansions but that further development of the assay is needed to make it fully useful.
Product Citations: 22
The TRX assay for triplet repeat expansions
Preprint on BioRxiv : the Preprint Server for Biology on 20 December 2024 by O’Brien, T., Salciute, K., et al.
-
FC/FACS
In Nature Genetics on 1 March 2024 by Mätlik, K., Baffuto, M., et al.
Brain region-specific degeneration and somatic expansions of the mutant Huntingtin (mHTT) CAG tract are key features of Huntington's disease (HD). However, the relationships among CAG expansions, death of specific cell types and molecular events associated with these processes are not established. Here, we used fluorescence-activated nuclear sorting (FANS) and deep molecular profiling to gain insight into the properties of cell types of the human striatum and cerebellum in HD and control donors. CAG expansions arise at mHTT in striatal medium spiny neurons (MSNs), cholinergic interneurons and cerebellar Purkinje neurons, and at mutant ATXN3 in MSNs from SCA3 donors. CAG expansions in MSNs are associated with higher levels of MSH2 and MSH3 (forming MutSβ), which can inhibit nucleolytic excision of CAG slip-outs by FAN1. Our data support a model in which CAG expansions are necessary but may not be sufficient for cell death and identify transcriptional changes associated with somatic CAG expansions and striatal toxicity.
© 2024. The Author(s).
-
Genetics
-
Neuroscience
In Nucleic Acids Research on 9 February 2024 by Li, Y., Zhang, Y., et al.
Common fragile sites (CFSs) are regions prone to chromosomal rearrangements, thereby contributing to tumorigenesis. Under replication stress (RS), CFSs often harbor under-replicated DNA regions at the onset of mitosis, triggering homology-directed repair known as mitotic DNA synthesis (MiDAS) to complete DNA replication. In this study, we identified an important role of DNA mismatch repair protein MutSβ (MSH2/MSH3) in facilitating MiDAS and maintaining CFS stability. Specifically, we demonstrated that MutSβ is required for the increased mitotic recombination induced by RS or FANCM loss at CFS-derived AT-rich and structure-prone sequences (CFS-ATs). We also found that MSH3 exhibits synthetic lethality with FANCM. Mechanistically, MutSβ is required for homologous recombination (HR) especially when DNA double-strand break (DSB) ends contain secondary structures. We also showed that upon RS, MutSβ is recruited to Flex1, a specific CFS-AT, in a PCNA-dependent but MUS81-independent manner. Furthermore, MutSβ interacts with RAD52 and promotes RAD52 recruitment to Flex1 following MUS81-dependent fork cleavage. RAD52, in turn, recruits XPF/ERCC1 to remove DNA secondary structures at DSB ends, enabling HR/break-induced replication (BIR) at CFS-ATs. We propose that the specific requirement of MutSβ in processing DNA secondary structures at CFS-ATs underlies its crucial role in promoting MiDAS and maintaining CFS integrity.
© The Author(s) 2023. Published by Oxford University Press on behalf of Nucleic Acids Research.
-
WB
-
Homo sapiens (Human)
-
Biochemistry and Molecular biology
-
Genetics
CNOT6: A Novel Regulator of DNA Mismatch Repair.
In Cells on 2 February 2022 by Song, P., Liu, S., et al.
DNA mismatch repair (MMR) is a highly conserved pathway that corrects both base-base mispairs and insertion-deletion loops (IDLs) generated during DNA replication. Defects in MMR have been linked to carcinogenesis and drug resistance. However, the regulation of MMR is poorly understood. Interestingly, CNOT6 is one of four deadenylase subunits in the conserved CCR4-NOT complex and it targets poly(A) tails of mRNAs for degradation. CNOT6 is overexpressed in acute lymphoblastic leukemia (ALL), acute myeloid leukemia (AML) and androgen-independent prostate cancer cells, which suggests that an altered expression of CNOT6 may play a role in tumorigenesis. Here, we report that a depletion of CNOT6 sensitizes human U2OS cells to N-methyl-N'nitro-N-nitrosoguanidine (MNNG) and leads to enhanced apoptosis. We also demonstrate that the depletion of CNOT6 upregulates MMR and decreases the mutation frequency in MMR-proficient cells. Furthermore, the depletion of CNOT6 increases the stability of mRNA transcripts from MMR genes, leading to the increased expression of MMR proteins. Our work provides insight into a novel CNOT6-dependent mechanism for regulating MMR.
-
Cell Biology
-
Genetics
In Cell Reports on 31 August 2021 by Goold, R., Hamilton, J., et al.
CAG repeat expansion in the HTT gene drives Huntington's disease (HD) pathogenesis and is modulated by DNA damage repair pathways. In this context, the interaction between FAN1, a DNA-structure-specific nuclease, and MLH1, member of the DNA mismatch repair pathway (MMR), is not defined. Here, we identify a highly conserved SPYF motif at the N terminus of FAN1 that binds to MLH1. Our data support a model where FAN1 has two distinct functions to stabilize CAG repeats. On one hand, it binds MLH1 to restrict its recruitment by MSH3, thus inhibiting the assembly of a functional MMR complex that would otherwise promote CAG repeat expansion. On the other hand, it promotes accurate repair via its nuclease activity. These data highlight a potential avenue for HD therapeutics in attenuating somatic expansion.
Copyright © 2021 The Author(s). Published by Elsevier Inc. All rights reserved.
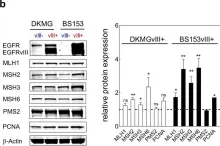
In Oncogene on 1 April 2020 by Struve, N., Binder, Z. A., et al.
Fig.4.B

-
WB
-
Collected and cropped from Oncogene by CiteAb, provided under a CC-BY license
Image 1 of 1