

The cells with compromised BRCA1 or BRCA2 (BRCA1/2) function accumulate stalled replication forks, which leads to replication-associated DNA damage and genomic instability, a signature of BRCA1/2-mutated tumours. Targeted therapies against BRCA1/2-mutated tumours exploit this vulnerability by introducing additional DNA lesions. Because homologous recombination (HR) repair is abrogated in the absence of BRCA1 or BRCA2, these lesions are specifically lethal to tumour cells, but not to the healthy tissue. Ligands that bind and stabilise G-quadruplexes (G4s) have recently emerged as a class of compounds that selectively eliminate the cells and tumours lacking BRCA1 or BRCA2. Pyridostatin is a small molecule that binds G4s and is specifically toxic to BRCA1/2-deficient cells in vitro. However, its in vivo potential has not yet been evaluated. Here, we demonstrate that pyridostatin exhibits a high specific activity against BRCA1/2-deficient tumours, including patient-derived xenograft tumours that have acquired PARP inhibitor (PARPi) resistance. Mechanistically, we demonstrate that pyridostatin disrupts replication leading to DNA double-stranded breaks (DSBs) that can be repaired in the absence of BRCA1/2 by canonical non-homologous end joining (C-NHEJ). Consistent with this, chemical inhibitors of DNA-PKcs, a core component of C-NHEJ kinase activity, act synergistically with pyridostatin in eliminating BRCA1/2-deficient cells and tumours. Furthermore, we demonstrate that pyridostatin triggers cGAS/STING-dependent innate immune responses when BRCA1 or BRCA2 is abrogated. Paclitaxel, a drug routinely used in cancer chemotherapy, potentiates the in vivo toxicity of pyridostatin. Overall, our results demonstrate that pyridostatin is a compound suitable for further therapeutic development, alone or in combination with paclitaxel and DNA-PKcs inhibitors, for the benefit of cancer patients carrying BRCA1/2 mutations.
© 2022 The Authors. Published under the terms of the CC BY 4.0 license.
Product Citations: 4
Anti-tumoural activity of the G-quadruplex ligand pyridostatin against BRCA1/2-deficient tumours.
In EMBO Molecular Medicine on 7 March 2022 by Groelly, F. J., Porru, M., et al.
-
WB
-
Biochemistry and Molecular biology
-
Cancer Research
PTEN Regulates Nonhomologous End Joining By Epigenetic Induction of NHEJ1/XLF.
In Molecular Cancer Research on 1 August 2018 by Sulkowski, P. L., Scanlon, S. E., et al.
DNA double-strand breaks (DSB) are the most cytotoxic DNA lesions, and up to 90% of DSBs require repair by nonhomologous end joining (NHEJ). Functional and genomic analyses of patient-derived melanomas revealed that PTEN loss is associated with NHEJ deficiency. In PTEN-null melanomas, PTEN complementation rescued the NHEJ defect; conversely, suppression of PTEN compromised NHEJ. Mechanistic studies revealed that PTEN promotes NHEJ through direct induction of expression of XRCC4-like factor (NHEJ1/XLF), which functions in DNA end bridging and ligation. PTEN was found to occupy the NHEJ1 gene promoter and to recruit the histone acetyltransferases, PCAF and CBP, inducing XLF expression. This recruitment activity was found to be independent of its phosphatase activity, but dependent on K128, a site of regulatory acetylation on PTEN. These findings define a novel function for PTEN in regulating NHEJ DSB repair, and therefore may assist in the design of individualized strategies for cancer therapy.Implications: PTEN is the second most frequently lost tumor suppressor gene. Here it is demonstrated that PTEN has a direct and novel regulatory role in NHEJ, a key DNA repair pathway in response to radiation and chemotherapy. Mol Cancer Res; 16(8); 1241-54. ©2018 AACR.©2018 American Association for Cancer Research.
-
WB
-
Cancer Research
-
Genetics
In Molecular Cancer Research on 1 April 2016 by Czochor, J. R., Sulkowski, P., et al.
miR-155 is an oncogenic miRNA that is often overexpressed in cancer and is associated with poor prognosis. miR-155 can target several DNA repair factors, including RAD51, MLH1, and MSH6, and its overexpression results in an increased mutation frequency in vitro, although the mechanism has yet to be fully understood. Here, we demonstrate that overexpression of miR-155 drives an increased mutation frequency both in vitro and in vivo, promoting genomic instability by affecting multiple DNA repair pathways. miR-155 overexpression causes a decrease in homologous recombination, but yields a concurrent increase in the error-prone nonhomologous end-joining pathway. Despite repressing established targets MLH1 and MSH6, the identified mutation pattern upon miR-155 overexpression does not resemble that of a mismatch repair-deficient background. Further investigation revealed that all four subunits of polymerase delta, a high-fidelity DNA replication, and repair polymerase are downregulated at the mRNA level in the context of miR-155 overexpression. FOXO3a, a transcription factor and known target of miR-155, has one or more putative binding site(s) in the promoter of all four polymerase delta subunits. Finally, suppression of FOXO3a by miR-155 or by siRNA knockdown is sufficient to repress the expression of the catalytic subunit of polymerase delta, POLD1, at the protein level, indicating that FOXO3a contributes to the regulation of polymerase delta levels.Taken together, miR-155 overexpression drives an increase in mutation frequency via multifaceted impact on DNA damage response and DNA repair pathways.©2016 American Association for Cancer Research.
-
WB
-
Cancer Research
In PLoS Genetics on 1 August 2014 by Terasawa, M., Shinohara, A., et al.
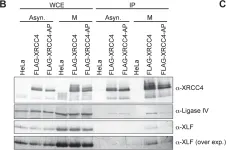
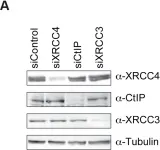
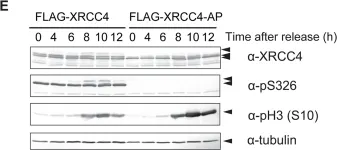
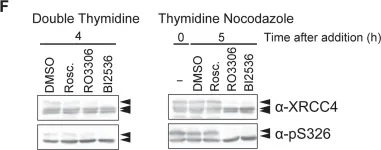
DNA double-strand breaks (DSBs) can be repaired by one of two major pathways-non-homologous end-joining (NHEJ) and homologous recombination (HR)-depending on whether cells are in G1 or S/G2 phase, respectively. However, the mechanisms of DSB repair during M phase remain largely unclear. In this study, we demonstrate that transient treatment of M-phase cells with the chemotherapeutic topoisomerase inhibitor etoposide induced DSBs that were often associated with anaphase bridge formation and genome instability such as dicentric chromosomes. Although most of the DSBs were carried over into the next G1 phase, some were repaired during M phase. Both NHEJ and HR, in particular NHEJ, promoted anaphase-bridge formation, suggesting that these repair pathways can induce genome instability during M phase. On the other hand, C-terminal-binding protein interacting protein (CtIP) suppressed anaphase bridge formation, implying that CtIP function prevents genome instability during mitosis. We also observed M-phase-specific phosphorylation of XRCC4, a regulatory subunit of the ligase IV complex specialized for NHEJ. This phosphorylation required cyclin-dependent kinase (CDK) activity as well as polo-like kinase 1 (Plk1). A phosphorylation-defective XRCC4 mutant showed more efficient M-phase DSB repair accompanied with an increase in anaphase bridge formation. These results suggest that phosphorylation of XRCC4 suppresses DSB repair by modulating ligase IV function to prevent genome instability during M phase. Taken together, our results indicate that XRCC4 is required not only for the promotion of NHEJ during interphase but also for its M-phase-specific suppression of DSB repair.
-
WB
-
Homo sapiens (Human)
-
Cell Biology
-
Genetics
In PLoS Genet on 1 August 2014 by Terasawa, M., Shinohara, A., et al.
Fig.4.C

-
WB
-
Homo sapiens (Human)
Collected and cropped from PLoS Genetics by CiteAb, provided under a CC-BY license
Image 1 of 6
In PLoS Genet on 1 August 2014 by Terasawa, M., Shinohara, A., et al.
Fig.5.B
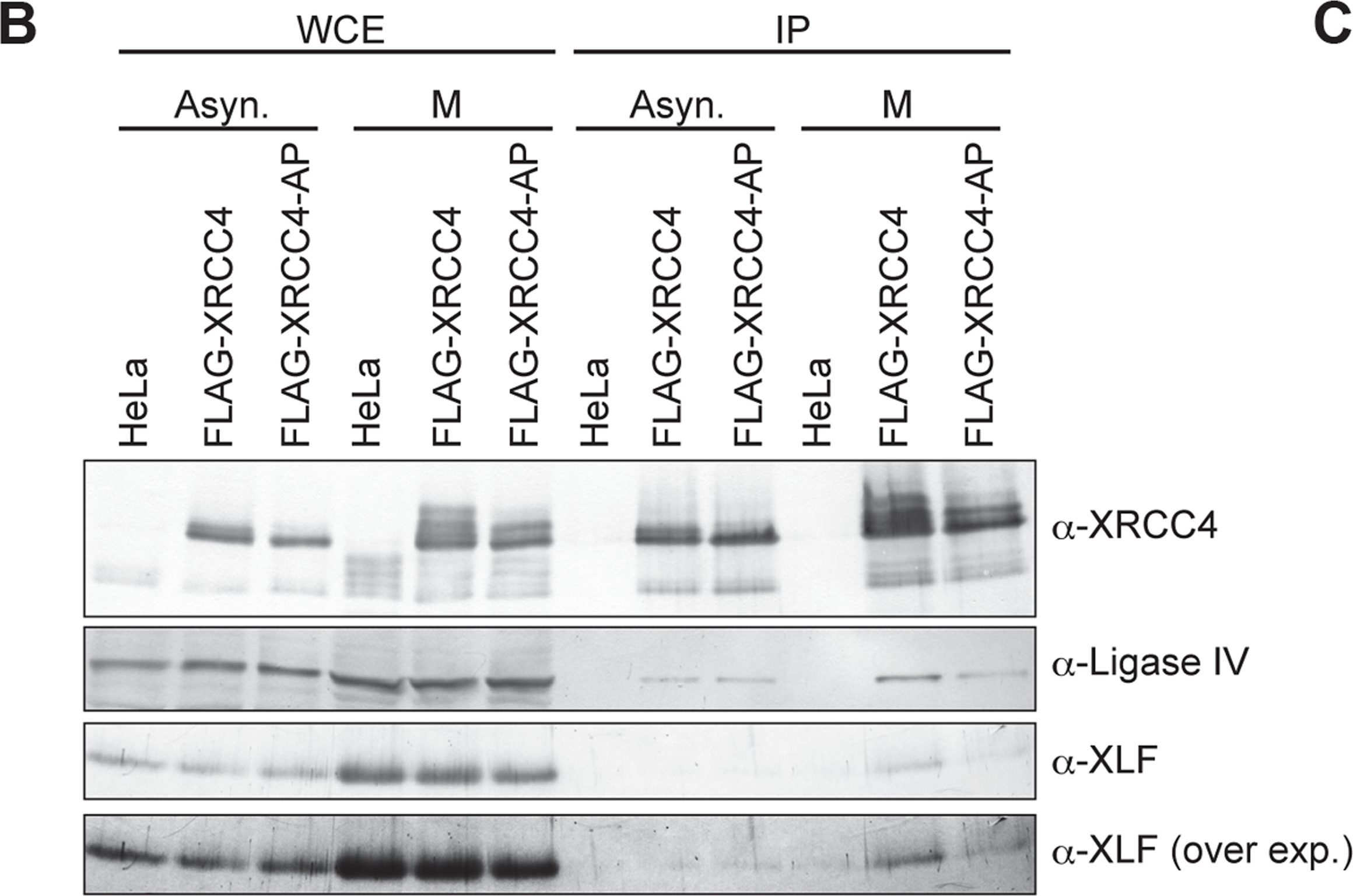
-
WB
-
Homo sapiens (Human)
Collected and cropped from PLoS Genetics by CiteAb, provided under a CC-BY license
Image 1 of 6
In PLoS Genet on 1 August 2014 by Terasawa, M., Shinohara, A., et al.
Fig.3.A
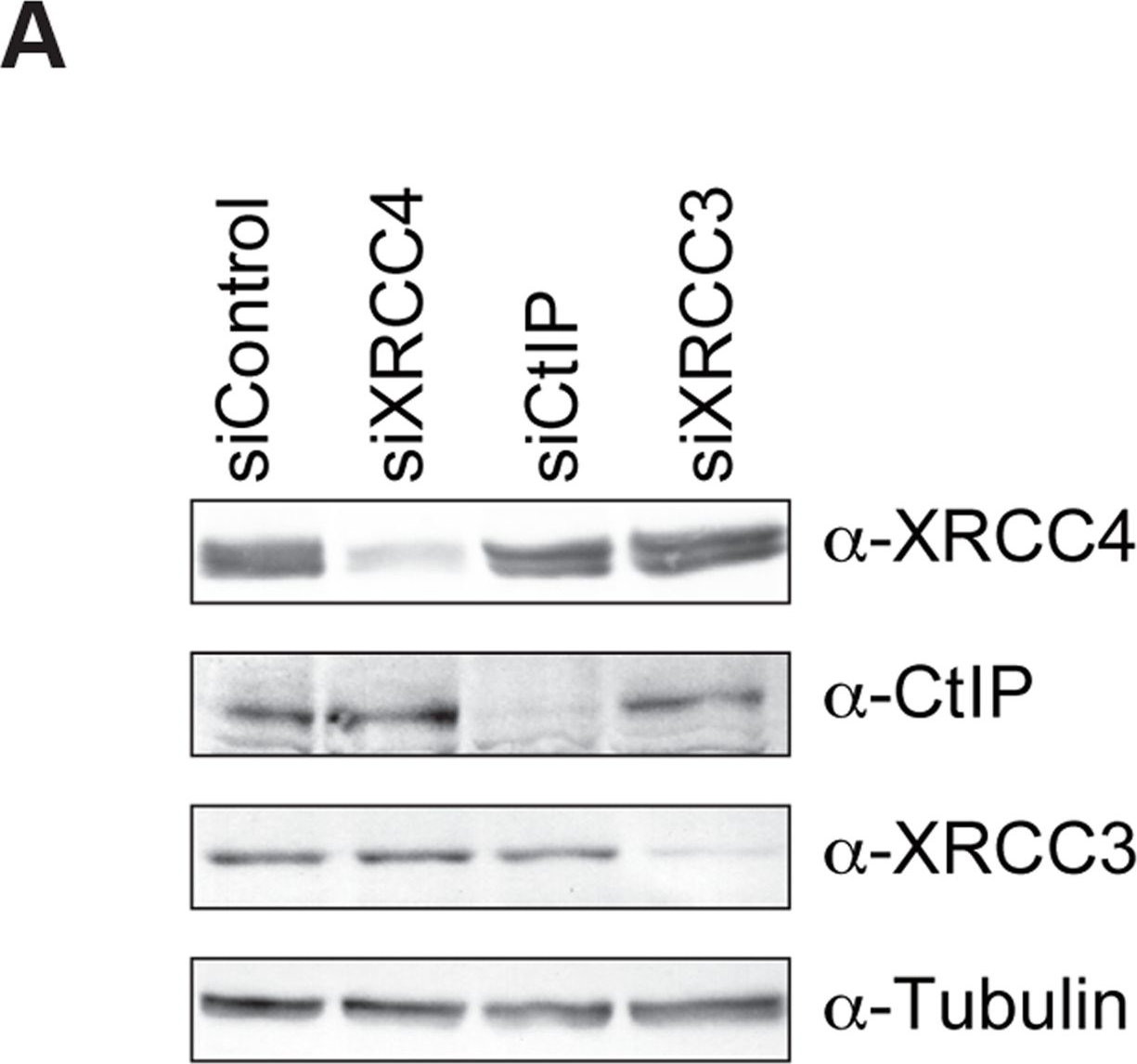
-
WB
-
Homo sapiens (Human)
Collected and cropped from PLoS Genetics by CiteAb, provided under a CC-BY license
Image 1 of 6
In PLoS Genet on 1 August 2014 by Terasawa, M., Shinohara, A., et al.
Fig.4.D

-
WB
-
Homo sapiens (Human)
Collected and cropped from PLoS Genetics by CiteAb, provided under a CC-BY license
Image 1 of 6
In PLoS Genet on 1 August 2014 by Terasawa, M., Shinohara, A., et al.
Fig.4.E
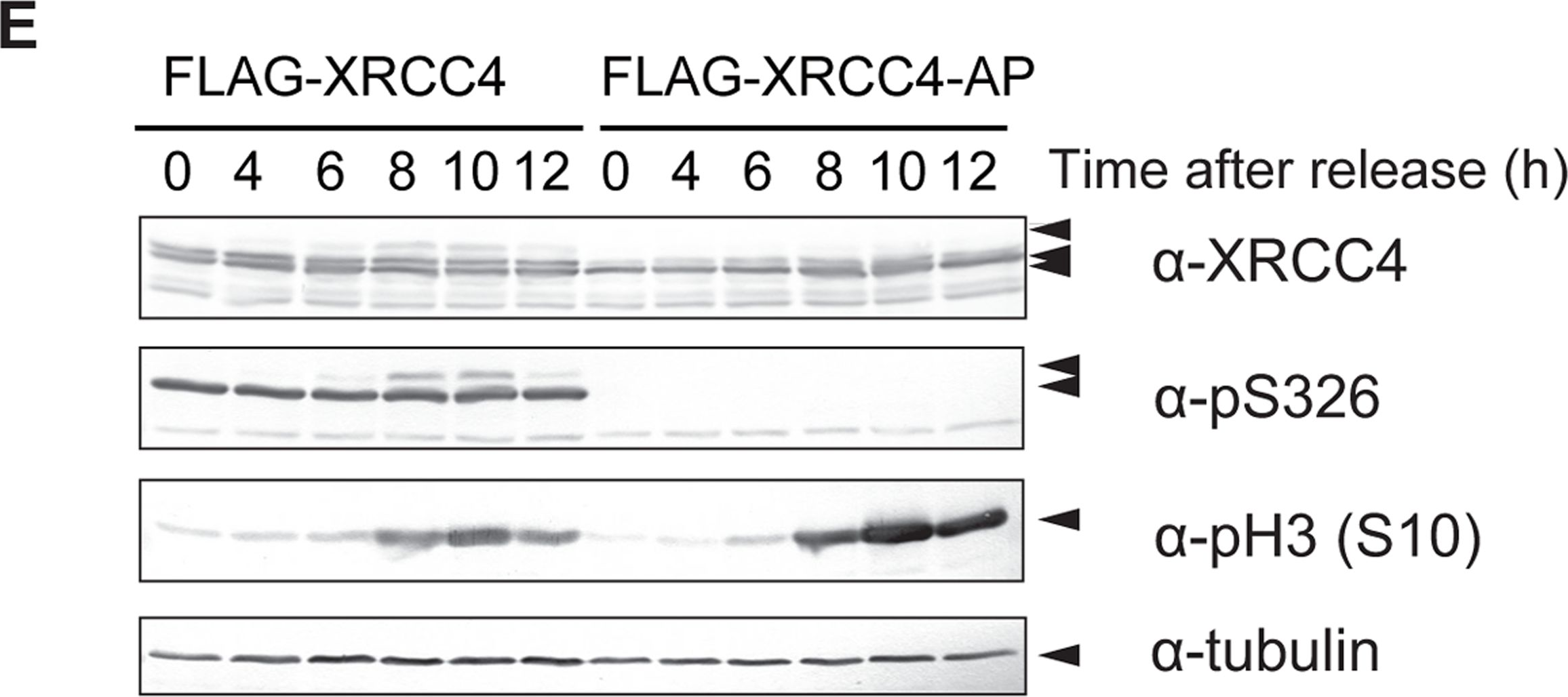
-
WB
-
Homo sapiens (Human)
Collected and cropped from PLoS Genetics by CiteAb, provided under a CC-BY license
Image 1 of 6
In PLoS Genet on 1 August 2014 by Terasawa, M., Shinohara, A., et al.
Fig.4.F
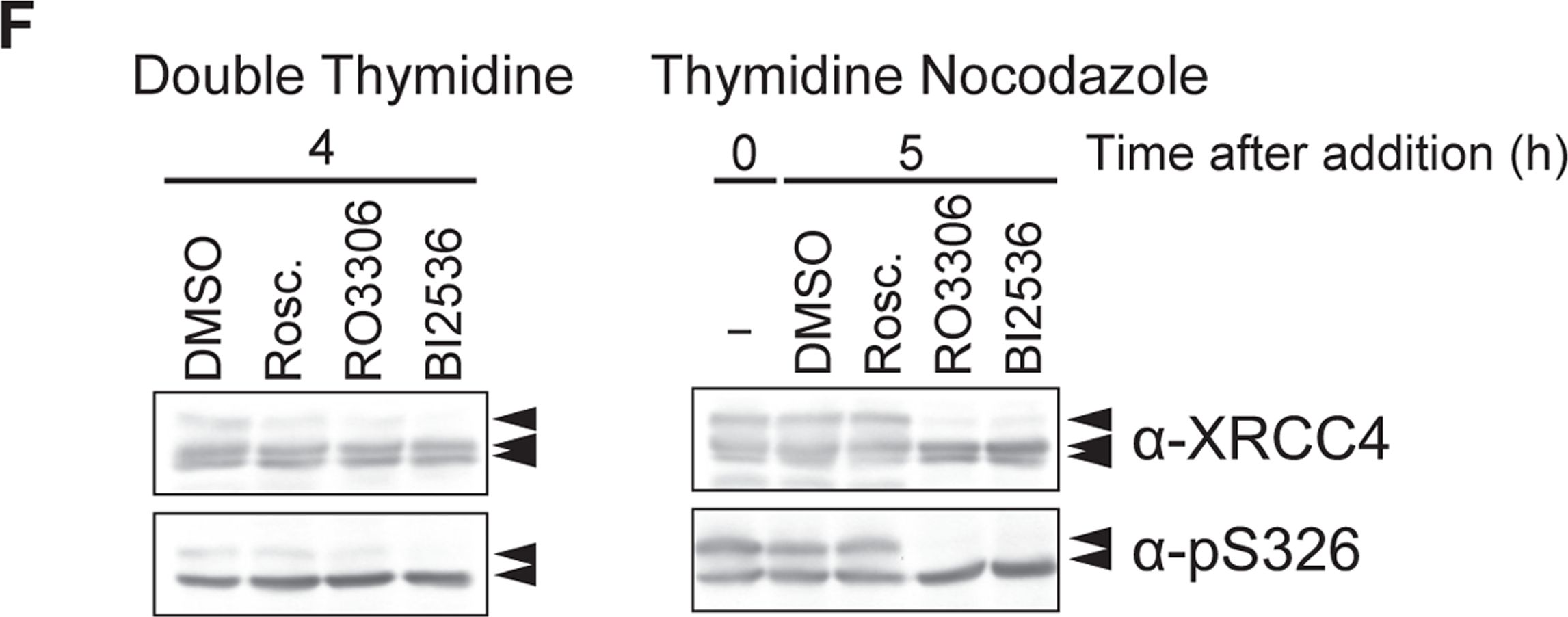
-
WB
-
Homo sapiens (Human)
Collected and cropped from PLoS Genetics by CiteAb, provided under a CC-BY license
Image 1 of 6