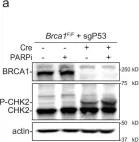
Microtubules and nuclear transmembrane SUN1/2 proteins promote the mobility of DNA Double Strand Breaks (DSBs) induced by ionizing radiation and the misrepair of one-ended DSBs induced in BRCA1-deficient cells by Poly(ADP-ribose) polymerase inhibitors (PARPi). However, whether microtubules promote aberrant DSBs repair by altering the nuclear structure and whether the nuclear structure itself plays a role in these processes is still unclear. Here we show that microtubule-dependent DSBs mobility in BRCA1-deficient cells after PARPi treatment is associated with nuclear envelope (NE) invaginations. Furthermore, increasing NE invaginations by Lmna deletion or inhibition of sphingolipid synthesis increases DSBs mobility, chromosomal aberrations, and PARPi cytotoxicity in BRCA1-deficient cells. These findings reveal a functional connection between the NE and DSB repair and suggest that drugs increasing NE deformability will enhance PARPi therapy efficacy in BRCA1-deficient cancers.
© 2025. The Author(s).
Product Citations: 34
In Nature Communications on 17 June 2025 by Faustini, E., Dello Stritto, A., et al.
-
WB
-
Mus musculus (House mouse)
-
Genetics
In Nature Cell Biology on 1 January 2025 by Hervé, S., Scelfo, A., et al.
Errors during cell division lead to aneuploidy, which is associated with genomic instability and cell transformation. In response to aneuploidy, cells activate the tumour suppressor p53 to elicit a surveillance mechanism that halts proliferation and promotes senescence. The molecular sensors that trigger this checkpoint are unclear. Here, using a tunable system of chromosome mis-segregation, we show that mitotic errors trigger nuclear deformation, nuclear softening, and lamin and heterochromatin alterations, leading to rapid p53/p21 activation upon mitotic exit in response to changes in nuclear mechanics. We identify mTORC2 and ATR as nuclear deformation sensors upstream of p53/p21 activation. While triggering mitotic arrest, the chromosome mis-segregation-induced alterations of nuclear envelope mechanics provide a fitness advantage for aneuploid cells by promoting nuclear deformation resilience and enhancing pro-invasive capabilities. Collectively, this work identifies a nuclear mechanical checkpoint triggered by altered chromatin organization that probably plays a critical role in cellular transformation and cancer progression.
© 2025. The Author(s).
-
Cell Biology
-
Genetics
UBE2D3 facilitates NHEJ by orchestrating ATM signalling through multi-level control of RNF168.
In Nature Communications on 12 June 2024 by Yalçin, Z., Lam, S. Y., et al.
Maintenance of genome integrity requires tight control of DNA damage response (DDR) signalling and repair, with phosphorylation and ubiquitination representing key elements. How these events are coordinated to achieve productive DNA repair remains elusive. Here we identify the ubiquitin-conjugating enzyme UBE2D3 as a regulator of ATM kinase-induced DDR that promotes non-homologous end-joining (NHEJ) at telomeres. UBE2D3 contributes to DDR-induced chromatin ubiquitination and recruitment of the NHEJ-promoting factor 53BP1, both mediated by RNF168 upon ATM activation. Additionally, UBE2D3 promotes NHEJ by limiting RNF168 accumulation and facilitating ATM-mediated phosphorylation of KAP1-S824. Mechanistically, defective KAP1-S824 phosphorylation and telomeric NHEJ upon UBE2D3-deficiency are linked to RNF168 hyperaccumulation and aberrant PP2A phosphatase activity. Together, our results identify UBE2D3 as a multi-level regulator of NHEJ that orchestrates ATM and RNF168 activities. Moreover, they reveal a negative regulatory circuit in the DDR that is constrained by UBE2D3 and consists of RNF168- and phosphatase-mediated restriction of KAP1 phosphorylation.
© 2024. The Author(s).
-
WB
DNA-PK controls Apollo's access to leading-end telomeres.
In Nucleic Acids Research on 8 May 2024 by Sonmez, C., Toia, B., et al.
The complex formed by Ku70/80 and DNA-PKcs (DNA-PK) promotes the synapsis and the joining of double strand breaks (DSBs) during canonical non-homologous end joining (c-NHEJ). In c-NHEJ during V(D)J recombination, DNA-PK promotes the processing of the ends and the opening of the DNA hairpins by recruiting and/or activating the nuclease Artemis/DCLRE1C/SNM1C. Paradoxically, DNA-PK is also required to prevent the fusions of newly replicated leading-end telomeres. Here, we describe the role for DNA-PK in controlling Apollo/DCLRE1B/SNM1B, the nuclease that resects leading-end telomeres. We show that the telomeric function of Apollo requires DNA-PKcs's kinase activity and the binding of Apollo to DNA-PK. Furthermore, AlphaFold-Multimer predicts that Apollo's nuclease domain has extensive additional interactions with DNA-PKcs, and comparison to the cryo-EM structure of Artemis bound to DNA-PK phosphorylated on the ABCDE/Thr2609 cluster suggests that DNA-PK can similarly grant Apollo access to the DNA end. In agreement, the telomeric function of DNA-PK requires the ABCDE/Thr2609 cluster. These data reveal that resection of leading-end telomeres is regulated by DNA-PK through its binding to Apollo and its (auto)phosphorylation-dependent positioning of Apollo at the DNA end, analogous but not identical to DNA-PK dependent regulation of Artemis at hairpins.
© The Author(s) 2024. Published by Oxford University Press on behalf of Nucleic Acids Research.
-
WB
-
Biochemistry and Molecular biology
-
Genetics
In Redox Biology on 1 April 2024 by Li, C., Deng, C., et al.
Reactive oxygen species (ROS) play a pivotal role in macrophage-mediated acute inflammation. However, the precise molecular mechanism by which ROS regulate macrophage polarization remains unclear. Here, we show that ROS function as signaling molecules that regulate M1 macrophage polarization through ataxia-telangiectasia mutated (ATM) and cell cycle checkpoint kinase 2 (Chk2), vital effector kinases in the DNA damage response (DDR) signaling pathway. We further demonstrate that Chk2 phosphorylates PKM2 at the T95 and T195 sites, promoting glycolysis and facilitating macrophage M1 polarization. In addition, Chk2 activation increases the Chk2-dependent expression of p21, inducing cell cycle arrest for subsequent macrophage M1 polarization. Finally, Chk2-deficient mice infected with lipopolysaccharides (LPS) display a significant decrease in lung inflammation and M1 macrophage counts. Taken together, these results suggest that inhibiting the ROS-Chk2 axis can prevent the excessive inflammatory activation of macrophages, and this pathway can be targeted to develop a novel therapy for inflammation-associated diseases and expand our understanding of the pathophysiological functions of DDR in innate immunity.
Copyright © 2024 The Author(s). Published by Elsevier B.V. All rights reserved.
-
Biochemistry and Molecular biology
-
Cell Biology
-
Immunology and Microbiology
In Nat Commun on 17 June 2025 by Faustini, E., Dello Stritto, A., et al.
Fig.1.A

-
WB
-
Collected and cropped from Nat Commun by CiteAb, provided under a CC-BY license
Image 1 of 3
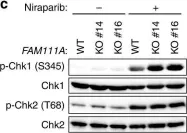
In Nat Commun on 12 March 2020 by Kojima, Y., Machida, Y., et al.
Fig.6.C

-
WB
-
Collected and cropped from Nat Commun by CiteAb, provided under a CC-BY license
Image 1 of 3
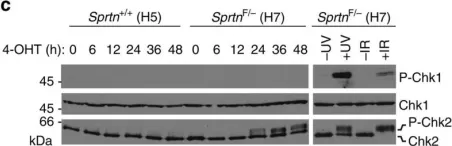
In Nat Commun on 11 December 2014 by Maskey, R. S., Kim, M. S., et al.
Fig.3.C

-
WB
-
Collected and cropped from Nat Commun by CiteAb, provided under a CC-BY license
Image 1 of 3