The transcriptional scaffolds C-terminal Binding Proteins (CtBP) 1 and 2 are overexpressed and act as oncogenic dependencies in multiple cancers but importantly encode a chemically targetable dehydrogenase domain. CtBP promotes survival of high grade serous ovarian carcinoma (HGSOC) cells by repressing expression of Death Receptors (DR) 4 and 5, which activate caspase 8-dependent apoptosis. We have previously developed a series of substrate competitive CtBP dehydrogenase inhibitors active in multiple cell and preclinical solid tumor models. In the current study, we validated CtBP 1 and 2 overexpression in a longitudinal series of primary and metastatic/recurrent HGSOC cases. Furthermore, our lead CtBP dehydrogenase inhibitor, JW-98 induced apoptosis and exhibited variable single agent IC 50 values in HGSOC cell lines, but depletion of nicotinamide adenine dinucleotide (NAD) using the NAD synthesis inhibitor GMX1778 strikingly sensitized tumor cells to JW-98 treatment. Mechanistically, the JW-98/GMX-1778 combination effectively disrupted CtBP dimerization that requires stoichiometric levels of intracellular NAD and is required for oncogenic transcriptional activities. Highlighting the translational potential of this combination, combined JW-98/GMX1778 treatment of OVCAR3 HGSOC xenografts in immunodeficient mice abrogated tumor growth without observable toxicity. CtBP/NAD combined inhibition represents a novel therapeutic strategy that could improve outcomes in chemoresistant HGSOC.
Product Citations: 53
Preprint on BioRxiv : the Preprint Server for Biology on 25 March 2025 by Chougoni, K. K., West, J., et al.
-
Cancer Research
The specificity of CTBP dehydrogenase inhibitors MTOB and 4-Cl-HIPP.
In MicroPublication. Biology on 19 February 2025 by Stickland, B. A., Greulich, F., et al.
C-terminal binding proteins (CTBPs) are conserved transcriptional repressors important in cancer and inflammation. Uniquely amongst transcriptional co-regulators, CTBPs possess a functional dehydrogenase domain. Since multiple malignancies display elevated CTBP levels, CTBP inhibitors targeting this dehydrogenase domain have been developed. While the importance of CTBPs dehydrogenase function for transcriptional regulation remains unclear, several studies have relied on CTBP inhibitors. In vitro experiments have confirmed binding of these compounds to CTBP's active site, however evidence for specificity is lacking. To address this, we treated wildtype and Ctbp1 , 2 double knockout J774.1 cells with MTOB or 4-Cl-HIPP and performed RNA-seq. We observed that both inhibitors elicit distinct transcriptional changes indicating non-overlapping modes of action. Moreover, the majority of changes induced by either inhibitor are observed in Ctbp1/2 double knockout cells suggesting off-target effects. We hypothesize that those CTBP dehydrogenase inhibitors lack specificity to CTBPs and emphasise careful revaluation of findings inferred from studies using those inhibitors.
Copyright: © 2025 by the authors.
Preprint on BioRxiv : the Preprint Server for Biology on 2 January 2025 by Pastoors, D., Havermans, M., et al.
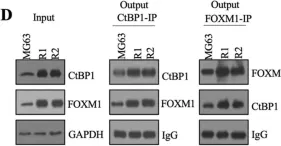
Enhancer translocations, due to 3q26 rearrangements, drive out-of-context MECOM expression in an aggressive subtype of acute myeloid leukemia (AML). Direct depletion of MECOM using an endogenous auxin-inducible degron immediately upregulates expression of myeloid differentiation factor CEBPA . MECOM depletion is also accompanied by a severe loss of stem cells and gain of differentiation. MECOM exerts its inhibitory effect by binding to the +42kb CEBPA enhancer, a gene essential for neutrophil development. This is partially dependent on the interaction between MECOM and its co-repressor CTBP2. We demonstrate that CEBPA overexpression can bypass the MECOM-mediated block of differentiation. In addition, AML patients with MECOM overexpression through enhancer hijacking show significantly reduced CEBPA . Our study directly connects two main players of myeloid transformation MECOM and CEBPA , and it provides insight into the mechanism by which MECOM maintains the stem cell state in a unique subtype of AML by inactivating CEBPA .
-
Cancer Research
In Nature Communications on 19 June 2024 by Goradia, N., Werner, S., et al.
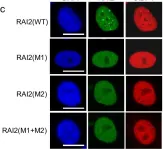
While the elucidation of regulatory mechanisms of folded proteins is facilitated due to their amenability to high-resolution structural characterization, investigation of these mechanisms in disordered proteins is more challenging due to their structural heterogeneity, which can be captured by a variety of biophysical approaches. Here, we used the transcriptional master corepressor CtBP, which binds the putative metastasis suppressor RAI2 through repetitive SLiMs, as a model system. Using cryo-electron microscopy embedded in an integrative structural biology approach, we show that RAI2 unexpectedly induces CtBP polymerization through filaments of stacked tetrameric CtBP layers. These filaments lead to RAI2-mediated CtBP nuclear foci and relieve its corepressor function in RAI2-expressing cancer cells. The impact of RAI2-mediated CtBP loss-of-function is illustrated by the analysis of a diverse cohort of prostate cancer patients, which reveals a substantial decrease in RAI2 in advanced treatment-resistant cancer subtypes. As RAI2-like SLiM motifs are found in a wide range of organisms, including pathogenic viruses, our findings serve as a paradigm for diverse functional effects through multivalent interaction-mediated polymerization by disordered proteins in healthy and diseased conditions.
© 2024. The Author(s).
-
WB
-
ICC-IF
-
Homo sapiens (Human)
Oncogene EVI1 drives acute myeloid leukemia via a targetable interaction with CTBP2.
In Science Advances on 17 May 2024 by Pastoors, D., Havermans, M., et al.
Acute myeloid leukemia (AML) driven by the activation of EVI1 due to chromosome 3q26/MECOM rearrangements is incurable. Because transcription factors such as EVI1 are notoriously hard to target, insight into the mechanism by which EVI1 drives myeloid transformation could provide alternative avenues for therapy. Applying protein folding predictions combined with proteomics technologies, we demonstrate that interaction of EVI1 with CTBP1 and CTBP2 via a single PLDLS motif is indispensable for leukemic transformation. A 4× PLDLS repeat construct outcompetes binding of EVI1 to CTBP1 and CTBP2 and inhibits proliferation of 3q26/MECOM rearranged AML in vitro and in xenotransplant models. This proof-of-concept study opens the possibility to target one of the most incurable forms of AML with specific EVI1-CTBP inhibitors. This has important implications for other tumor types with aberrant expression of EVI1 and for cancers transformed by different CTBP-dependent oncogenic transcription factors.
-
Cancer Research
In Nat Commun on 19 June 2024 by Goradia, N., Werner, S., et al.
Fig.2.C

-
ICC-IF
-
Collected and cropped from Nat Commun by CiteAb, provided under a CC-BY license
Image 1 of 17
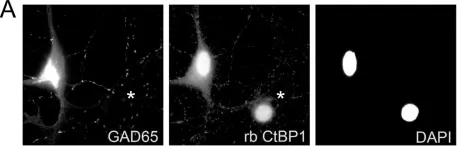
In Mol Neurobiol on 1 August 2023 by Saraiva, C., Lopes-Nunes, J., et al.
Fig.3.A

-
WB
-
Collected and cropped from Mol Neurobiol by CiteAb, provided under a CC-BY license
Image 1 of 17
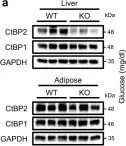
In Nat Commun on 2 November 2021 by Sekiya, M., Kainoh, K., et al.
Fig.5.A

-
WB
-
Collected and cropped from Nat Commun by CiteAb, provided under a CC-BY license
Image 1 of 17
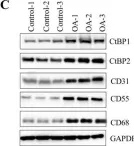
In J Cancer on 5 January 2021 by Chen, X., Zhang, Q., et al.
Fig.4.D

-
WB
-
Collected and cropped from J Cancer by CiteAb, provided under a CC-BY license
Image 1 of 17
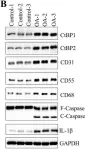
In Int J Biol Sci on 7 March 2020 by Sun, X., Xiao, L., et al.
Fig.1.C

-
WB
-
Collected and cropped from Int J Biol Sci by CiteAb, provided under a CC-BY license
Image 1 of 17
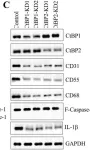
In Int J Biol Sci on 7 March 2020 by Sun, X., Xiao, L., et al.
Fig.3.B

-
WB
-
Collected and cropped from Int J Biol Sci by CiteAb, provided under a CC-BY license
Image 1 of 17
In Int J Biol Sci on 7 March 2020 by Sun, X., Xiao, L., et al.
Fig.3.C

-
WB
-
Collected and cropped from Int J Biol Sci by CiteAb, provided under a CC-BY license
Image 1 of 17
In Int J Biol Sci on 7 March 2020 by Sun, X., Xiao, L., et al.
Fig.5.A

-
WB
-
Collected and cropped from Int J Biol Sci by CiteAb, provided under a CC-BY license
Image 1 of 17
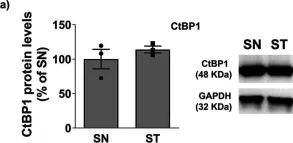
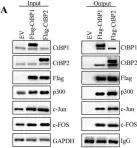
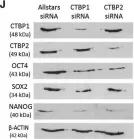
In Stem Cell Reports on 9 April 2019 by Arthur, S. A., Blaydes, J. P., et al.
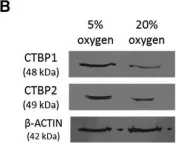
Fig.4.G

-
WB
-
Collected and cropped from Stem Cell Reports by CiteAb, provided under a CC-BY license
Image 1 of 17
In Stem Cell Reports on 9 April 2019 by Arthur, S. A., Blaydes, J. P., et al.
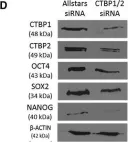
Fig.4.D

-
WB
-
Collected and cropped from Stem Cell Reports by CiteAb, provided under a CC-BY license
Image 1 of 17
In Stem Cell Reports on 9 April 2019 by Arthur, S. A., Blaydes, J. P., et al.
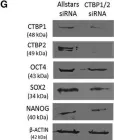
Fig.1.D

-
WB
-
Collected and cropped from Stem Cell Reports by CiteAb, provided under a CC-BY license
Image 1 of 17
In Stem Cell Reports on 9 April 2019 by Arthur, S. A., Blaydes, J. P., et al.
Fig.1.B

-
WB
-
Collected and cropped from Stem Cell Reports by CiteAb, provided under a CC-BY license
Image 1 of 17
In Stem Cell Reports on 9 April 2019 by Arthur, S. A., Blaydes, J. P., et al.
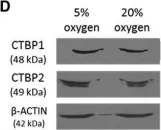
Fig.4.J

-
WB
-
Collected and cropped from Stem Cell Reports by CiteAb, provided under a CC-BY license
Image 1 of 17
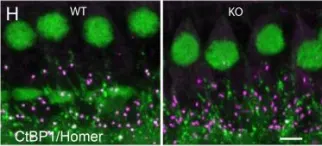
In Elife on 12 January 2018 by Becker, L., Schnee, M. E., et al.
Fig.1.H

-
IHC-IF
-
Mus musculus (House mouse)
Collected and cropped from Elife by CiteAb, provided under a CC-BY license
Image 1 of 17
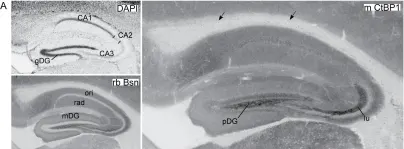
In PLoS One on 30 June 2012 by Hübler, D., Rankovic, M., et al.
Fig.5.A

-
ICC-IF
-
Collected and cropped from PLoS One by CiteAb, provided under a CC-BY license
Image 1 of 17
In PLoS One on 30 June 2012 by Hübler, D., Rankovic, M., et al.
Fig.3.A

-
IHC
-
Collected and cropped from PLoS One by CiteAb, provided under a CC-BY license
Image 1 of 17
In BMC Genomics on 27 December 2007 by Schulz, T. C., Swistowska, A. M., et al.
Fig.4.B

-
ICC-IF
-
Homo sapiens (Human)
Collected and cropped from BMC Genomics by CiteAb, provided under a CC-BY license
Image 1 of 17