Huntington's disease (HD) is caused by the expansion of the polyglutamine stretch in huntingtin protein (HTT) resulting in hallmark aggresomes/inclusion bodies (IBs) composed of mutant huntingtin protein (mHTT) and its fragments. Stimulating autophagy to enhance mHTT clearance is considered a potential therapeutic strategy for HD. Our recent evaluation of the autophagic-lysosomal pathway (ALP) in human HD brain reveals upregulated lysosomal biogenesis and relatively normal autophagy flux in early Vonsattel grade brains, but impaired autolysosome clearance in late grade brains, suggesting that autophagy stimulation could have therapeutic benefits as an early clinical intervention. Here, we tested this hypothesis by crossing the Q175 HD knock-in model with our autophagy reporter mouse TRGL (Thy-1-RFP-GFP-LC3) to investigate in vivo neuronal ALP dynamics. In the Q175 and/or TRGL/Q175 mice, mHTT was detected in autophagic vacuoles and also exhibited a high level of colocalization with autophagy receptors p62/SQSTM1 and ubiquitin in the IBs. Compared to the robust lysosomal pathology in late-stage human HD striatum, ALP alterations in Q175 models are also late-onset but milder, that included a lowered phospho-p70S6K level, lysosome depletion, and autolysosome elevation including more poorly acidified autolysosomes and larger-sized lipofuscin granules, reflecting impaired autophagic flux. Administration of a mTOR inhibitor to 6-mo-old TRGL/Q175 normalized lysosome number, ameliorated aggresome pathology while reducing mHTT-, p62-, and ubiquitin-immunoreactivities, suggesting the beneficial potential of autophagy modulation at early stages of disease progression.
© 2025, Stavrides et al.
Product Citations: 42
In eLife on 20 May 2025 by Stavrides, P., Goulbourne, C. N., et al.
-
WB
-
Cell Biology
A primary cilia-autophagy axis in hippocampal neurons is essential to maintain cognitive resilience.
In Nature Aging on 1 March 2025 by Rivagorda, M., Romeo-Guitart, D., et al.
Blood-borne factors are essential to maintain neuronal synaptic plasticity and cognitive resilience throughout life. One such factor is osteocalcin (OCN), a hormone produced by osteoblasts that influences multiple physiological processes, including hippocampal neuronal homeostasis. However, the mechanism through which this blood-borne factor communicates with neurons remains unclear. Here we show the importance of a core primary cilium (PC) protein-autophagy axis in mediating the effects of OCN. We found that the OCN receptor GPR158 is present at the PC of hippocampal neurons and mediates the regulation of autophagy machinery by OCN. During aging, autophagy and PC core proteins are reduced in neurons, and restoring their levels is sufficient to improve cognitive impairments in aged mice. Mechanistically, the induction of this axis by OCN is dependent on the PC-dependent cAMP response element-binding protein signaling pathway. Altogether, this study demonstrates that the PC-autophagy axis is a gateway to mediate communication between blood-borne factors and neurons, and it advances understanding of the mechanisms involved in age-related cognitive decline.
© 2025. The Author(s).
-
WB
-
Cell Biology
-
Neuroscience
Preprint on BioRxiv : the Preprint Server for Biology on 30 May 2024 by Stavrides, P., Goulbourne, C. N., et al.
A bstract Huntington’s disease (HD) is caused by expansion of the polyglutamine stretch in huntingtin protein (HTT) resulting in hallmark aggresomes/inclusion bodies (IBs) composed of mutant huntingtin protein (mHTT) and its fragments. Stimulating autophagy to enhance mHTT clearance is considered a potential therapeutic strategy for HD. Our recent evaluation of the autophagic-lysosomal pathway (ALP) in human HD brain reveals upregulated lysosomal biogenesis and relatively normal autophagy flux in early Vonsattel grade brains, but impaired autolysosome clearance in late grade brains, suggesting that autophagy stimulation could have therapeutic benefits as an earlier clinical intervention. Here, we tested this hypothesis by crossing the Q175 HD knock-in model with our autophagy reporter mouse TRGL ( T hy-1- R FP- G FP- L C3) to investigate in vivo neuronal ALP dynamics. In the Q175 and/or TRGL/Q175 mice, mHTT was detected in autophagic vacuoles and also exhibited high level colocalization with autophagy receptors p62/SQSTM1 and ubiquitin in the IBs. Compared to the robust lysosomal pathology in late-stage human HD striatum, ALP alterations in Q175 models are also late-onset but milder that included a lowered phospho-p70S6K level, lysosome depletion and autolysosome elevation including more poorly acidified autolysosomes and larger-sized lipofuscin granules, reflecting impaired autophagic flux. Administration of a mTOR inhibitor to 6-mo-old TRGL/Q175 normalized lysosome number, ameliorated aggresome pathology while reducing mHTT-, p62- and ubiquitin-immunoreactivities, suggesting beneficial potential of autophagy modulation at early stages of disease progression.
-
Mus musculus (House mouse)
-
Cell Biology
In Heliyon on 15 January 2024 by Das, N., Mukherjee, S., et al.
Epidemiological as well as experimental studies have established that the pineal hormone melatonin has inhibitory effects on different types of cancers. Several mechanisms have been proposed for the anticancer activities of melatonin, but the fundamental molecular pathways still require clarity. We developed a mouse model of breast cancer using Ehrlich's ascites carcinoma (injected in the 4th mammary fat pad of female Swiss albino mice) and investigated the possibility of targeting the autophagy-inflammation-EMT colloquy to restrict breast tumor progression using melatonin as intervention. Contrary to its conventional antioxidant role, melatonin was shown to augment intracellular ROS and initiate ROS-dependent apoptosis in our system, by modulating the p53/JNK & NF-κB/pJNK expressions/interactions. Melatonin-induced ROS promoted SIRT1 activity. Interplay between SIRT1 and NF-κB/p65 is known to play a pivotal role in regulating the crosstalk between autophagy and inflammation. Persistent inflammation in the tumor microenvironment and subsequent activation of the IL-6/STAT3/NF-κB feedback loop promoted EMT and suppression of autophagy through activation of PI3K/Akt/mTOR signaling pathway. Melatonin disrupted NF-κB/SIRT1 interactions blocking IL-6/STAT3/NF-κB pathway. This led to reversal of pro-inflammatory bias in the breast tumor microenvironment and augmented autophagic responses. The interactions between p62/Twist1, NF-κB/Beclin1 and NF-κB/Slug were altered by melatonin to strike a balance between autophagy, inflammation and EMT, leading to tumor regression. This study provides critical insights into how melatonin could be utilized in treating breast cancer via inhibition of the PI3K/Akt/mTOR signaling and differential modulation of SIRT1 and NF-κB proteins, leading to the establishment of apoptotic and autophagic fates in breast cancer cells.
© 2023 The Authors.
-
Mus musculus (House mouse)
-
Cancer Research
-
Cell Biology
-
Immunology and Microbiology
In Cellular and Molecular Life Sciences : CMLS on 19 August 2022 by Carvalho, C., Correia, S. C., et al.
Diabetes has been associated with an increased risk of cognitive decline and dementia. However, the mechanisms underlying this association remain unclear and no effective therapeutic interventions exist. Accumulating evidence demonstrates that mitochondrial defects are a key feature of diabetes contributing to neurodegenerative events. It has also been demonstrated that the putative tumor suppressor WW domain-containing oxidoreductase 1 (WWOX) can interact with mitochondria in several pathological conditions. However, its role in diabetes-associated neurodegeneration remains unknown. So, this study aimed to evaluate the role of WWOX activation in high glucose-induced neuronal damage and death. Our experiments were mainly performed in differentiated SH-SY5Y neuroblastoma cells exposed to high glucose and treated (or not) with Zfra1-31, the specific inhibitor of WWOX. Several parameters were analyzed namely cell viability, WWOX activation (tyrosine 33 residue phosphorylation), mitochondrial function, reactive oxygen species (ROS) production, biogenesis, and dynamics, autophagy and oxidative stress/damage. The levels of the neurotoxic proteins amyloid β (Aβ) and phosphorylated Tau (pTau) and of synaptic integrity markers were also evaluated. We observed that high glucose increased the levels of activated WWOX. Interestingly, brain cortical and hippocampal homogenates from young (6-month old) diabetic GK rats showed increased levels of activated WWOX compared to older GK rats (12-month old) suggesting that WWOX plays an early role in the diabetic brain. In neuronal cells, high glucose impaired mitochondrial respiration, dynamics and biogenesis, increased mitochondrial ROS production and decreased mitochondrial membrane potential and ATP production. More, high glucose augmented oxidative stress/damage and the levels of Aβ and pTau proteins and affected autophagy, contributing to the loss of synaptic integrity and cell death. Of note, the activation of WWOX preceded mitochondrial dysfunction and cell death. Importantly, the inhibition of WWOX with Zfra1-31 reversed, totally or partially, the alterations promoted by high glucose. Altogether our observations demonstrate that under high glucose conditions WWOX activation contributes to mitochondrial anomalies and neuronal damage and death, which suggests that WWOX is a potential therapeutic target for early interventions. Our findings also support the efficacy of Zfra1-31 in treating hyperglycemia/diabetes-associated neurodegeneration.
© 2022. The Author(s), under exclusive licence to Springer Nature Switzerland AG.
-
Biochemistry and Molecular biology
-
Cell Biology
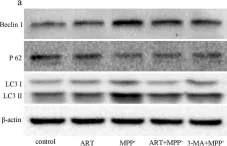
In J Biol Res (Thessalon) on 25 February 2021 by Yan, J., Ma, H., et al.
Fig.5.A

-
WB
-
Collected and cropped from J Biol Res (Thessalon) by CiteAb, provided under a CC-BY license
Image 1 of 9
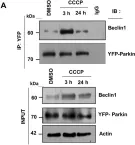
In Sci Rep on 12 January 2018 by Kumar, A. & Shaha, C.
Fig.4.A

-
WB
-
Homo sapiens (Human)
Collected and cropped from Sci Rep by CiteAb, provided under a CC-BY license
Image 1 of 9
In Sci Rep on 12 January 2018 by Kumar, A. & Shaha, C.
Fig.5.B

-
WB
-
Homo sapiens (Human)
Collected and cropped from Sci Rep by CiteAb, provided under a CC-BY license
Image 1 of 9
In Sci Rep on 12 January 2018 by Kumar, A. & Shaha, C.
Fig.6.B

-
WB
-
Homo sapiens (Human)
Collected and cropped from Sci Rep by CiteAb, provided under a CC-BY license
Image 1 of 9
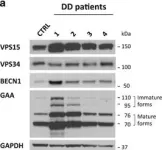
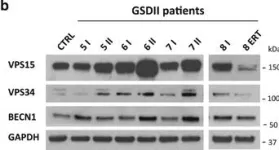
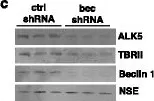
In Cell Death Dis on 19 January 2017 by Nascimbeni, A. C., Fanin, M., et al.
Fig.3.A
-
WB
-
Homo sapiens (Human)
Collected and cropped from Cell Death Dis by CiteAb, provided under a CC-BY license
Image 1 of 9
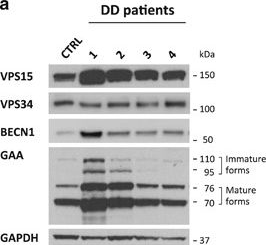
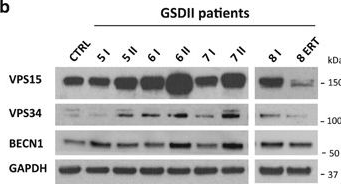
In Cell Death Dis on 19 January 2017 by Nascimbeni, A. C., Fanin, M., et al.
Fig.3.B
-
WB
-
Homo sapiens (Human)
Collected and cropped from Cell Death Dis by CiteAb, provided under a CC-BY license
Image 1 of 9
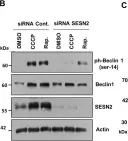
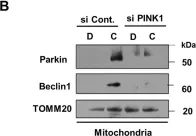
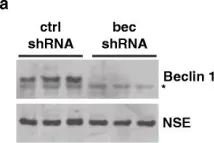
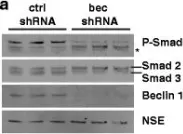
In Mol Neurodegener on 21 December 2015 by O'Brien, C. E., Bonanno, L., et al.
Fig.1.A

-
WB
-
Homo sapiens (Human)
Collected and cropped from Mol Neurodegener by CiteAb, provided under a CC-BY license
Image 1 of 9
In Mol Neurodegener on 21 December 2015 by O'Brien, C. E., Bonanno, L., et al.
Fig.7.A

-
WB
-
Mus musculus (House mouse)
Collected and cropped from Mol Neurodegener by CiteAb, provided under a CC-BY license
Image 1 of 9
In Mol Neurodegener on 21 December 2015 by O'Brien, C. E., Bonanno, L., et al.
Fig.3.C

-
WB
-
Homo sapiens (Human)
Collected and cropped from Mol Neurodegener by CiteAb, provided under a CC-BY license
Image 1 of 9