
Contact inhibition of locomotion (CIL) is a process that regulates cell motility upon collision with other cells. Improper regulation of CIL has been implicated in cancer cell dissemination. Here, we identify the cell adhesion molecule JAM-A as a central regulator of CIL in tumor cells. JAM-A is part of a multimolecular signaling complex in which tetraspanins CD9 and CD81 link JAM-A to αvβ5 integrin. JAM-A binds Csk and inhibits the activity of αvβ5 integrin-associated Src. Loss of JAM-A results in increased activities of downstream effectors of Src, including Erk1/2, Abi1, and paxillin, as well as increased activity of Rac1 at cell-cell contact sites. As a consequence, JAM-A-depleted cells show increased motility, have a higher cell-matrix turnover, and fail to halt migration when colliding with other cells. We also find that proper regulation of CIL depends on αvβ5 integrin engagement. Our findings identify a molecular mechanism that regulates CIL in tumor cells and have implications on tumor cell dissemination.
© 2022 Kummer et al.
Product Citations: 8
A JAM-A-tetraspanin-αvβ5 integrin complex regulates contact inhibition of locomotion.
In The Journal of Cell Biology on 4 April 2022 by Kummer, D., Steinbacher, T., et al.
-
WB
-
Cell Biology
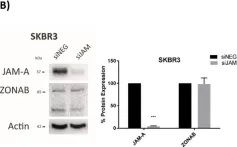
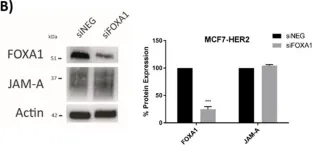
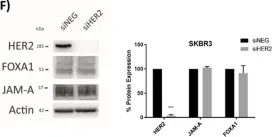
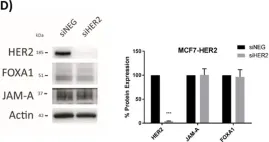
A Transcriptional Link between HER2, JAM-A and FOXA1 in Breast Cancer.
In Cells on 19 February 2022 by Cruz, R. G. B., Madden, S. F., et al.
Overexpression of the human epidermal growth factor receptor-2 (HER2) is associated with aggressive disease in breast and certain other cancers. At a cellular level, the adhesion protein Junctional Adhesion Molecule-A (JAM-A) has been reported to regulate the expression of HER3 via a transcriptional pathway involving FOXA1. Since FOXA1 is also a suggested transcription factor for HER2, this study set out to determine if JAM-A regulates HER2 expression via a similar mechanism. An integrated tripartite approach was taken, involving cellular expression studies after targeted disruption of individual players in the putative pathway, in silico identification of relevant HER2 promoter regions and, finally, interrogation of cancer patient survival databases to deconstruct functionally important links between HER2, JAM-A and FOXA1 gene expression. The outcome of these investigations revealed a unidirectional pathway in which JAM-A expression transcriptionally regulates that of HER2 by influencing the binding of FOXA1 to a specific site in the HER2 gene promoter. Moreover, a correlation between JAM-A and HER2 gene expression was identified in 75% of a sample of 40 cancer types from The Cancer Genome Atlas, and coincident high mean mRNA expression of JAM-A, HER2 and FOXA1 was associated with poorer survival outcomes in HER2-positive (but not HER2-negative) patients with either breast or gastric tumors. These investigations provide the first evidence of a transcriptional pathway linking JAM-A, HER2 and FOXA1 in cancer settings, and support potential future pharmacological targeting of JAM-A as an upstream regulator of HER2.
-
WB
-
Homo sapiens (Human)
-
Biochemistry and Molecular biology
-
Cancer Research
-
Cell Biology
In Cancers on 19 February 2021 by Cruz, R. G. B., Madden, S. F., et al.
The success of breast cancer therapies targeting the human epidermal growth factor receptor-2 (HER2) is limited by the development of drug resistance by mechanisms including upregulation of HER3. Having reported that HER2 expression and resistance to HER2-targeted therapies can be regulated by Junctional Adhesion Molecule-A (JAM-A), this study investigated if JAM-A regulates HER3 expression. Expressional alteration of JAM-A in breast cancer cells was used to test expressional effects on HER3 and its effectors, alongside associated functional behaviors, in vitro and semi-in vivo. HER3 transcription factors were identified and tested for regulation by JAM-A. Finally a patient tissue microarray was used to interrogate connections between putative pathway components connecting JAM-A and HER3. This study reveals for the first time that HER3 and its effectors are regulated at gene/protein expression level by JAM-A in breast cancer cell lines; with functional consequences in in vitro and semi-in vivo models. In bioinformatic, cellular and patient tissue models, this was associated with regulation of the HER3 transcription factor FOXA1 by JAM-A via a pathway involving β-catenin. Our data suggest a novel model whereby JAM-A expression regulates β-catenin localization, in turn regulating FOXA1 expression, which could drive HER3 gene transcription. JAM-A merits investigation as a novel target to prevent upregulation of HER3 during the development of resistance to HER2-targeted therapies, or to reduce HER3-dependent tumorigenic signaling.
-
WB
-
Biochemistry and Molecular biology
-
Cancer Research
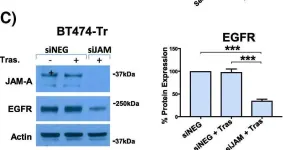
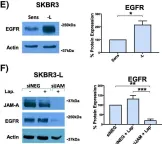
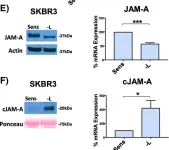
In Breast Cancer Research : BCR on 20 November 2018 by Leech, A. O., Vellanki, S. H., et al.
Junctional adhesion molecule-A (JAM-A) is an adhesion molecule whose overexpression on breast tumor tissue has been associated with aggressive cancer phenotypes, including human epidermal growth factor receptor-2 (HER2)-positive disease. Since JAM-A has been described to regulate HER2 expression in breast cancer cells, we hypothesized that JAM-dependent stabilization of HER2 could participate in resistance to HER2-targeted therapies.
Using breast cancer cell line models resistant to anti-HER2 drugs, we investigated JAM-A expression and the effect of JAM-A silencing on biochemical/functional parameters. We also tested whether altered JAM-A expression/processing underpinned differences between drug-sensitive and -resistant cells and acted as a biomarker of patients who developed resistance to HER2-targeted therapies.
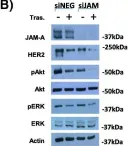
Silencing JAM-A enhanced the anti-proliferative effects of anti-HER2 treatments in trastuzumab- and lapatinib-resistant breast cancer cells and further reduced HER2 protein expression and Akt phosphorylation in drug-treated cells. Increased epidermal growth factor receptor expression observed in drug-resistant models was normalized upon JAM-A silencing. JAM-A was highly expressed in all of a small cohort of HER2-positive patients whose disease recurred following anti-HER2 therapy. High JAM-A expression also correlated with metastatic disease at the time of diagnosis in another patient cohort resistant to trastuzumab therapy. Importantly, cleavage of JAM-A was increased in drug-resistant cell lines in conjunction with increased expression of ADAM-10 and -17 metalloproteases. Pharmacological inhibition or genetic silencing studies suggested a particular role for ADAM-10 in reducing JAM-A cleavage and partially re-sensitizing drug-resistant cells to the anti-proliferative effects of HER2-targeted drugs. Functionally, recombinant cleaved JAM-A enhanced breast cancer cell invasion in vitro and both invasion and proliferation in a semi-in vivo model. Finally, cleaved JAM-A was detectable in the serum of a small cohort of HER2-positive patients and correlated significantly with resistance to HER2-targeted therapy.
Collectively, our data suggest a novel model whereby increased expression and cleavage of JAM-A drive tumorigenic behavior and act as a biomarker and potential therapeutic target for resistance to HER2-targeted therapies.
-
WB
-
Cancer Research
In Cellular Signalling on 1 December 2017 by Datta, A., Sandilands, E., et al.
The formation of lumens in epithelial tissues requires apical-basal polarization of cells, and the co-ordination of this individual polarity collectively around a contiguous lumen. Signals from the Extracellular Matrix (ECM) instruct epithelia as to the orientation of where basal, and thus consequently apical, surfaces should be formed. We report that this pathway is normally absent in Calu-3 human lung adenocarcinoma cells in 3-Dimensional culture, but that paracrine signals from MRC5 lung fibroblasts can induce correct orientation of polarity and acinar morphogenesis. We identify HGF, acting through the c-Met receptor, as the key polarity-inducing morphogen, which acts to activate β1-integrin-dependent adhesion. HGF and ECM-derived integrin signals co-operate via a c-Src-dependent inhibition of the RhoA-ROCK1 signalling pathway via p190A RhoGAP. This occurred via controlling localization of these signalling pathways to the ECM-abutting surface of cells in 3-Dimensional culture. Thus, stromal derived signals can influence morphogenesis in epithelial cells by controlling activation and localization of cell polarity pathways.
Copyright © 2017 Elsevier Inc. All rights reserved.
-
Homo sapiens (Human)
-
Cancer Research
-
Cell Biology
In Cells on 19 February 2022 by Cruz, R. G. B., Madden, S. F., et al.
Fig.3.B

-
WB
-
Collected and cropped from Cells by CiteAb, provided under a CC-BY license
Image 1 of 9
In Cells on 19 February 2022 by Cruz, R. G. B., Madden, S. F., et al.
Fig.6.B

-
WB
-
Collected and cropped from Cells by CiteAb, provided under a CC-BY license
Image 1 of 9
In Cells on 19 February 2022 by Cruz, R. G. B., Madden, S. F., et al.
Fig.6.F

-
WB
-
Collected and cropped from Cells by CiteAb, provided under a CC-BY license
Image 1 of 9
In Cells on 19 February 2022 by Cruz, R. G. B., Madden, S. F., et al.
Fig.6.D

-
WB
-
Collected and cropped from Cells by CiteAb, provided under a CC-BY license
Image 1 of 9
In Breast Cancer Res on 20 November 2018 by Leech, A. O., Vellanki, S. H., et al.
Fig.1.B

-
WB
-
Collected and cropped from Breast Cancer Res by CiteAb, provided under a CC-BY license
Image 1 of 9
In Breast Cancer Res on 20 November 2018 by Leech, A. O., Vellanki, S. H., et al.
Fig.1.E

-
WB
-
Collected and cropped from Breast Cancer Res by CiteAb, provided under a CC-BY license
Image 1 of 9
In Breast Cancer Res on 20 November 2018 by Leech, A. O., Vellanki, S. H., et al.
Fig.2.C

-
WB
-
Collected and cropped from Breast Cancer Res by CiteAb, provided under a CC-BY license
Image 1 of 9
In Breast Cancer Res on 20 November 2018 by Leech, A. O., Vellanki, S. H., et al.
Fig.2.E

-
WB
-
Collected and cropped from Breast Cancer Res by CiteAb, provided under a CC-BY license
Image 1 of 9
In Breast Cancer Res on 20 November 2018 by Leech, A. O., Vellanki, S. H., et al.
Fig.3.E

-
WB
-
Collected and cropped from Breast Cancer Res by CiteAb, provided under a CC-BY license
Image 1 of 9