The NEIL3 DNA glycosylase is a base excision repair enzyme that excises bulky base lesions from DNA. Although NEIL3 has been shown to unhook interstrand crosslinks (ICL) in Xenopus extracts, how NEIL3 participants in ICL repair in human cells and its corporation with the canonical Fanconi anemia (FA)/BRCA pathway remain unclear. Here we show that the NEIL3 and the FA/BRCA pathways are non-epistatic in psoralen-ICL repair. The NEIL3 pathway is the major pathway for repairing psoralen-ICL, and the FA/BRCA pathway is only activated when NEIL3 is not present. Mechanistically, NEIL3 is recruited to psoralen-ICL in a rapid, PARP-dependent manner. Importantly, the NEIL3 pathway repairs psoralen-ICLs without generating double-strand breaks (DSBs), unlike the FA/BRCA pathway. In addition, we found that the RUVBL1/2 complex physically interact with NEIL3 and function within the NEIL3 pathway in psoralen-ICL repair. Moreover, TRAIP is important for the recruitment of NEIL3 but not FANCD2, and knockdown of TRAIP promotes FA/BRCA pathway activation. Interestingly, TRAIP is non-epistatic with both NEIL3 and FA pathways in psoralen-ICL repair, suggesting that TRAIP may function upstream of the two pathways. Taken together, the NEIL3 pathway is the major pathway to repair psoralen-ICL through a unique DSB-free mechanism in human cells.
© The Author(s) 2020. Published by Oxford University Press on behalf of Nucleic Acids Research.
Product Citations: 7
Cooperation of the NEIL3 and Fanconi anemia/BRCA pathways in interstrand crosslink repair.
In Nucleic Acids Research on 6 April 2020 by Li, N., Wang, J., et al.
-
WB
-
Biochemistry and Molecular biology
Reptin drives tumour progression and resistance to chemotherapy in nonsmall cell lung cancer.
In The European Respiratory Journal on 1 July 2018 by Mikesch, J. H., Schwammbach, D., et al.
While targeted nonsmall cell lung cancer (NSCLC) therapies have improved the outcome of defined disease subtypes, prognosis for most patients remains poor. We found the AAA+ ATPase Reptin to be highly expressed in the vast majority of 278 NSCLC tumour samples. Thus, the objective of the study was to assess the role of Reptin in NSCLC.Survival analyses of 1145 NSCLC patients revealed that high RNA expression levels of Reptin are associated with adverse outcome. Knockdown of Reptin in human NSCLC cells impaired growth ex vivo and eliminated engraftment in a xenograft model. Reptin directly interacted with histone deacetylase 1 (HDAC1) as the critical mechanism driving NSCLC tumour progression. Pharmacological disruption of the Reptin/HDAC1 complex resulted in a substantial decrease in NSCLC cell proliferation and induced significant sensitisation to cisplatin.Our results identify Reptin as a novel independent prognostic factor and as a key regulator mediating proliferation and clonal growth of human NSCLC cells ex vivo and in vivo We unveil a Reptin/HDAC1 protein complex whose pharmacological disruption sensitises NSCLC cells to cisplatin, suggesting this approach for application in clinical trials.
Copyright ©ERS 2018.
-
Cancer Research
In Cell Biochemistry and Function on 1 August 2017 by Raymond, A. A., Javary, J., et al.
Hepatocellular carcinoma (HCC) is the main primary cancer of the liver. Many studies have shown that insulin resistance is a risk factor for HCC. We previously discovered the overexpression and oncogenic role of the Reptin/RUVBL2 ATPase in HCC. Here, we found that Reptin silencing enhanced insulin sensitivity in 2 HCC cell lines, as shown by a large potentiation of insulin-induced AKT phosphorylation on Ser473 and Thr308, and of downstream signalling. Reptin silencing did not affect the tyrosine phosphorylation of the insulin receptor nor of IRS1, but it enhanced the tyrosine phosphorylation of the p85 subunit of PI3K. The expression of the SHP-1/PTPN6 phosphatase, which dephosphorylates p85, was reduced after Reptin depletion. Forced expression of SHP-1 restored a normal AKT phosphorylation after insulin treatment in cells where Reptin was silenced, demonstrating that the downregulation of SHP1 is mechanistically linked to increased Akt phosphorylation. In conclusion, we have uncovered a new function for Reptin in regulating insulin signalling in HCC cells via the regulation of SHP-1 expression. We suggest that the regulation of insulin sensitivity by Reptin contributes to its oncogenic action in the liver.
Copyright © 2017 John Wiley & Sons, Ltd.
-
Biochemistry and Molecular biology
-
Cancer Research
-
Endocrinology and Physiology
Reptin regulates DNA double strand breaks repair in human hepatocellular carcinoma.
In PLoS ONE on 16 April 2015 by Raymond, A. A., Benhamouche, S., et al.
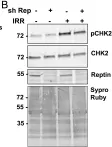
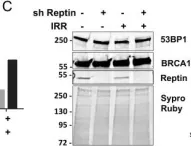
Reptin/RUVBL2 is overexpressed in most hepatocellular carcinomas and is required for the growth and viability of HCC cells. Reptin is involved in several chromatin remodeling complexes, some of which are involved in the detection and repair of DNA damage, but data on Reptin involvement in the repair of DNA damage are scarce and contradictory. Our objective was to study the effects of Reptin silencing on the repair of DNA double-strand breaks (DSB) in HCC cells. Treatment of HuH7 cells with etoposide (25 μM, 30 min) or γ irradiation (4 Gy) increased the phosphorylation of H2AX by 1.94 ± 0.13 and 2.0 ± 0.02 fold, respectively. These values were significantly reduced by 35 and 65 % after Reptin silencing with inducible shRNA. Irradiation increased the number of BRCA1 (3-fold) and 53BP1 foci (7.5 fold). Depletion of Reptin reduced these values by 62 and 48%, respectively. These defects in activation and/or recruitment of repair proteins were not due to a decreased number of DSBs as measured by the COMET assay. All these results were confirmed in the Hep3B cell line. Protein expression of ATM and DNA-PKcs, the major H2AX kinases, was significantly reduced by 52 and 61 % after Reptin depletion whereas their mRNA level remained unchanged. Phosphorylation of Chk2, another ATM target, was not significantly altered. Using co-immunoprecipitation, we showed an interaction between Reptin and DNA-PKcs. The half-life of newly-synthesized DNA-PKcs was reduced when Reptin was silenced. Finally, depletion of Reptin was synergistic with etoposide or γ irradiation to reduce cell growth and colony formation. In conclusion, Reptin is an important cofactor for the repair of DSBs. Our data, combined with those of the literature suggests that it operates at least in part by regulating the expression of DNA-PKcs by a stabilization mechanism. Overexpression of Reptin in HCC could be a factor of resistance to treatment, consistent with the observed overexpression of Reptin in subgroups of chemo-resistant breast and ovarian cancers.
-
WB
-
Homo sapiens (Human)
-
Cancer Research
-
Genetics
In Cancer Science on 1 January 2012 by Izumi, N., Yamashita, A., et al.
Heat shock protein 90 (Hsp90), a conserved molecular chaperone for a specific set of proteins critical for signal transduction including several oncogenic proteins, has been recognized as a promising target for anticancer therapy. Hsp90 inhibition also sensitizes cancer cells to DNA damage. However, the underlying mechanisms are not fully understood. Here, we provide evidence that Hsp90 is a general regulator of phosphatidylinositol 3-kinase-related protein kinase (PIKK) family proteins, central regulators of stress responses including DNA damage. Inhibition of Hsp90 causes a reduction of all PIKK and suppresses PIKK-mediated signaling. In addition, Hsp90 forms complexes with RUVBL1/2 complex and Tel2 complex, both of which have been shown to interact with all PIKK and control their abundance and functions. These results suggest that Hsp90 can form multiple complexes with the RUVBL1/2 complex and Tel2 complex and function in the regulation of PIKK, providing additional rationale for the effectiveness of Hsp90 inhibition for anticancer therapy, including sensitization to DNA damage.
© 2011 Japanese Cancer Association.
-
Cancer Research
In PLoS One on 16 April 2015 by Raymond, A. A., Benhamouche, S., et al.
Fig.5.B

-
WB
-
Homo sapiens (Human)
Collected and cropped from PLoS One by CiteAb, provided under a CC-BY license
Image 1 of 2
In PLoS One on 16 April 2015 by Raymond, A. A., Benhamouche, S., et al.
Fig.2.C

-
WB
-
Homo sapiens (Human)
Collected and cropped from PLoS One by CiteAb, provided under a CC-BY license
Image 1 of 2