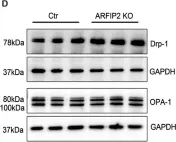

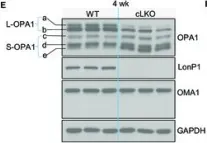
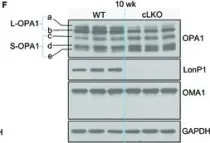
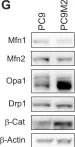

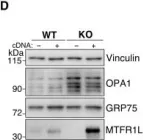
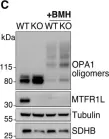
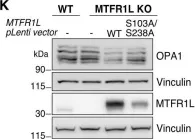
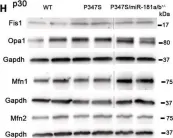
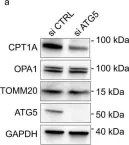
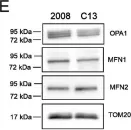
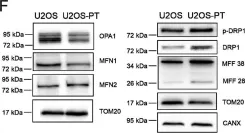
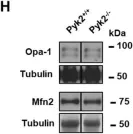

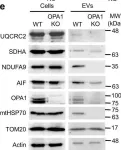
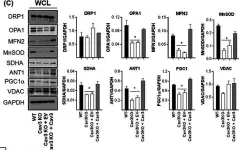

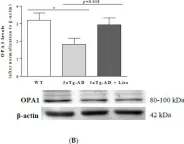

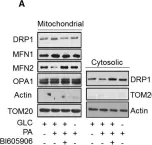
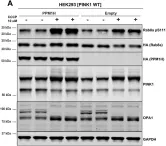
Recent studies have highlighted the importance of mitochondria in NP cells and articular chondrocyte health. Since the understanding of mechanisms governing mitochondrial dynamics in these tissues is lacking, we investigated the role of OPA1, a mitochondrial fusion protein, in their homeostasis. OPA1 knockdown in NP cells altered mitochondrial size and cristae shape and increased the oxygen consumption rate. OPA1 governed the morphology of multiple organelles, including peroxisomes, early endosomes and cis-Golgi and loss resulted in the dysregulation of autophagy. Metabolic profiling and 13C-flux analyses revealed TCA cycle anaplerosis and altered metabolism in OPA1-deficient NP cells. Noteworthy, Opa1AcanCreERT2 mice showed age-dependent disc degeneration, osteoarthritis, and vertebral osteopenia. RNA-Sequencing of Opa1cKO NP tissue revealed dysregulation of metabolism, autophagy, cytoskeletal reorganization, and extracellular matrix and shared strong thematic similarities with a subset of human degenerative NP samples. Our findings underscore that maintenance of mitochondrial dynamics and multi-organelle cross-talk is critical in preserving metabolic homeostasis of disc and cartilage.
© 2025. The Author(s).
Product Citations: 142
The loss of OPA1 accelerates intervertebral disc degeneration and osteoarthritis in aged mice.
In Nature Communications on 1 July 2025 by Madhu, V., Meadows, M. H., et al.
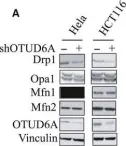
The MFN2 Q367H variant reveals a novel pathomechanism connected to mtDNA-mediated inflammation.
In Life Science Alliance on 1 June 2025 by Zaman, M., Sharma, G., et al.
Pathogenic variants in the mitochondrial protein MFN2 are typically associated with a peripheral neuropathy phenotype, but can also cause a variety of additional pathologies including myopathy. Here, we identified an uncharacterized MFN2 variant, Q367H, in a patient diagnosed with late-onset distal myopathy, but without peripheral neuropathy. Supporting the hypothesis that this variant contributes to the patient's pathology, patient fibroblasts and transdifferentiated myoblasts showed changes consistent with impairment of several MFN2 functions. We also observed mtDNA outside of the mitochondrial network that colocalized with early endosomes, and measured activation of both TLR9 and cGAS-STING inflammation pathways that sense mtDNA. Re-expressing the Q367H variant in MFN2 KO cells also induced mtDNA release, demonstrating this phenotype is a direct result of the variant. As elevated inflammation can cause myopathy, our findings linking the Q367H MFN2 variant with elevated TLR9 and cGAS-STING signalling can explain the patient's myopathy. Thus, we characterize a novel MFN2 variant in a patient with an atypical presentation that separates peripheral neuropathy and myopathy phenotypes, and establish a potential pathomechanism connecting MFN2 dysfunction to mtDNA-mediated inflammation.
© 2025 Zaman et al.
-
WB
-
Homo sapiens (Human)
-
Immunology and Microbiology
In Journal of Translational Medicine on 24 March 2025 by Wang, R., Wang, J., et al.
Pulmonary arterial hypertension (PAH) is a chronic disorder characterized by the excessive proliferation of pulmonary arterial smooth muscle cells (PASMCs). Recent studies indicate that Mitochondrial fusion protein 2 (Mfn2) maintains intracellular calcium (Ca2+) homeostasis via the mitochondria-associated endoplasmic reticulum membranes (MAMs) pathway, thereby inhibiting PASMCs proliferation and reducing pulmonary artery pressure. However, the precise mechanisms remain unclear.
This study explored the roles of Mfn2 and IP3R3 in PAH progression by assessing their expression in lung tissues of a monocrotaline (MCT)-induced PAH rat model. Immunoprecipitation assays were performed to confirm the interaction between Mfn2 and IP3R3. PASMCs were treated with either silenced or overexpressed Mfn2 and exposed to TNF-ɑ to observe effects on ER stress, IP3R3 expression, mitochondrial Ca2+ transport, and mitochondrial integrity. We also evaluated the effects of 4-phenylbutyric acid (4-PBA) and cistanche phenylethanol glycosides (CPGs) on the Mfn2-IP3R3 interaction in a TNF-α-induced PAH cell model, focusing on Ca2+ transport and mitochondrial structure.
Mfn2 expression was significantly down-regulated in the MCT-induced PAH rat model. Inhibition of ER stress upregulated Mfn2 expression, downregulated IP3R3 expression, increased mitochondrial Ca2+ concentration, and reduced autophagy, improving pulmonary hemodynamics and vascular remodeling. Overexpression of Mfn2 reduced ER stress, decreased IP3R3 expression, decreased mitochondrial Ca2+ transport, and restored mitochondrial integrity. Immunoprecipitation assays confirmed the interaction between Mfn2 and IP3R3. Inhibition of IP3R3 elevated Mfn2 levels, yielding similar beneficial effects as Mfn2 overexpression. 4-PBA and CPGs modulated the Mfn2-IP3R3 signaling axis, effectively inhibiting PAH progression.
Mfn2 mediates mitochondrial Ca2+ transport via IP3R3, suppressing PASMCs proliferation and pulmonary vascular remodeling, underscoring Mfn2's potential in regulating metabolic processes and vascular remodeling in PAH. These findings provide new insights for developing PAH-targeted therapeutics and establish a theoretical basis for traditional Chinese medicine in PAH prevention and treatment.
© 2025. The Author(s).
-
Cardiovascular biology
Inhibition of mitochondrial OMA1 ameliorates osteosarcoma tumorigenesis.
In Cell Death & Disease on 1 November 2024 by Chen, L., Chen, D., et al.
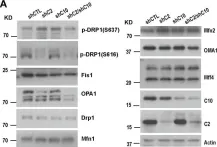
OMA1 is an ATP-independent zinc metalloprotease essential for maintaining mitochondrial homeostasis and plays a vital role in tumorigenesis. Depending on the type of cancer, a decrease in OMA1 expression has been linked to a varying prognosis for patients. The role of OMA1 in human osteosarcoma (OS), one of the most prevalent malignant bone tumors, remains elusive. Here, we observed elevated OMA1 expression in OS tumor tissues from four patients with advanced OS. Knockout of OMA1 in OS cells significantly reduces OS tumor weight and size, and lung metastatic nodules in BALB/c nude mice. Immunohistochemistry analysis showed a significant decrease in Ki67 and an increase in Cleaved-caspase 3 in OMA1 knockout tumor samples. Mechanistically, we found that OMA1 deficiency increases the levels of PINK1 and Parkin and consequently induces excessive mitophagy, leading to increased apoptosis and reduced cell proliferation and invasion in OS cells. Specifically, OMA1 deficiency reduces the amount of cytosolic p53 and p53-associated cytosolic Parkin but increases mitochondrial p53, which may lead to enhanced apoptosis. Regarding the effect on cell proliferation and invasion, loss of OMA1 reduces mitochondrial ROS levels and increases cytosolic glycogen synthase kinase 3β (GSK3β) levels, thereby increasing interaction between GSK3β and β-catenin and then reducing cytosolic and nuclear β-catenin. This contributes to reduced cell proliferation and migration in OMA1-deficient cells. Moreover, we found that ciclopirox (CPX), an antifungal drug, induces OMA1 self-cleavage and L-OMA1 degradation in cultured OS cells. CPX also reduces tumor development of control OS cells but not OMA1-deficient OS cells in mice. These findings strongly support the important role of OMA1 in OS tumorigenesis and suggest that OMA1 may be a valuable prognostic marker and a promising therapeutic target for OS.
© 2024. The Author(s).
-
Cancer Research
-
Cell Biology
In Scientific Reports on 29 October 2024 by Lin, Y., Cheng, W., et al.
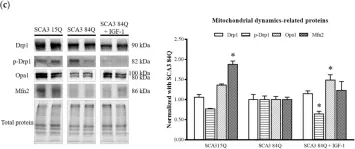
This study investigated the therapeutic effects of astragaloside IV (AST) on spinocerebellar ataxia type 3 (SCA3), also known as Machado-Joseph disease (MJD), a neurodegenerative disorder. Human neuroblastoma SK-N-SH cells expressing mutant ataxin-3 protein with 78 CAG repeats (MJD78) were employed as an in vitro model. Protein expression analysis demonstrated that AST treatment reduced mutant ataxin-3 protein expression and aggregation by enhancing the autophagic process in MJD78 cells. Elevated oxidative stress levels in MJD78 cells were significantly reduced following AST treatment, which also enhanced antioxidant capacity, as evidenced by flow cytometry and antioxidant enzyme activity assays. Furthermore, AST treatment ameliorated mitochondrial dysfunction in MJD78 cells, including improvements in mitochondrial membrane potential, respiration, and mitochondrial dynamics. In conclusion, AST administration increased antioxidant capacity, reduced both cellular and mitochondrial oxidative stress, and improved mitochondrial quality control processes through fusion, fission, and autophagy. These mechanisms collectively reduced intracellular mutant ataxin-3 protein aggregation, thereby achieving therapeutic efficacy in the SCA3 model.
© 2024. The Author(s).
-
WB
-
Cell Biology
In Antioxidants (Basel) on 8 January 2024 by Guo, H., Rogg, M., et al.
Fig.6.D

-
WB
-
Collected and cropped from Antioxidants (Basel) by CiteAb, provided under a CC-BY license
Image 1 of 41
In Dis Model Mech on 1 September 2023 by Millet, A. M. C., Coustham, C., et al.
Fig.2.A

-
WB
-
Collected and cropped from Dis Model Mech by CiteAb, provided under a CC-BY license
Image 1 of 41
In Research (Wash D C) on 19 June 2023 by Li, Y., Huang, D., et al.
Fig.2.E

-
WB
-
Collected and cropped from Research (Wash D C) by CiteAb, provided under a CC-BY license
Image 1 of 41
In Research (Wash D C) on 19 June 2023 by Li, Y., Huang, D., et al.
Fig.2.F

-
WB
-
Collected and cropped from Research (Wash D C) by CiteAb, provided under a CC-BY license
Image 1 of 41
In Cell Death Dis on 5 April 2023 by Noguchi, M., Kohno, S., et al.
Fig.1.G

-
WB
-
Collected and cropped from Cell Death Dis by CiteAb, provided under a CC-BY license
Image 1 of 41
In Cells on 21 March 2023 by Swirski, S., May, O., et al.
Fig.2.A

-
WB
-
Mus musculus (House mouse)
Collected and cropped from Cells by CiteAb, provided under a CC-BY license
Image 1 of 41
In Sci Adv on 11 November 2022 by Tilokani, L., Russell, F. M., et al.
Fig.3.D

-
WB
-
Collected and cropped from Sci Adv by CiteAb, provided under a CC-BY license
Image 1 of 41
In Sci Adv on 11 November 2022 by Tilokani, L., Russell, F. M., et al.
Fig.3.C

-
WB
-
Collected and cropped from Sci Adv by CiteAb, provided under a CC-BY license
Image 1 of 41
In Sci Adv on 11 November 2022 by Tilokani, L., Russell, F. M., et al.
Fig.4.K

-
WB
-
Collected and cropped from Sci Adv by CiteAb, provided under a CC-BY license
Image 1 of 41
In EMBO Mol Med on 8 November 2022 by Carrella, S., Di Guida, M., et al.
Fig.2.H

-
WB
-
Collected and cropped from EMBO Mol Med by CiteAb, provided under a CC-BY license
Image 1 of 41
In Nat Commun on 19 May 2022 by Mece, O., Houbaert, D., et al.
Fig.5.A

-
WB
-
Collected and cropped from Nat Commun by CiteAb, provided under a CC-BY license
Image 1 of 41
In Cell Death Dis on 22 April 2022 by Vianello, C., Cocetta, V., et al.
Fig.1.E

-
WB
-
Homo sapiens (Human)
Collected and cropped from Cell Death Dis by CiteAb, provided under a CC-BY license
Image 1 of 41
In Cell Death Dis on 22 April 2022 by Vianello, C., Cocetta, V., et al.
Fig.1.F

-
WB
-
Homo sapiens (Human)
Collected and cropped from Cell Death Dis by CiteAb, provided under a CC-BY license
Image 1 of 41
In Cells on 1 March 2022 by López-Molina, L., Fernández-Irigoyen, J., et al.
Fig.5.H

-
WB
-
Collected and cropped from Cells by CiteAb, provided under a CC-BY license
Image 1 of 41
In Biomedicines on 21 February 2022 by Lin, Y. S., Cheng, W. L., et al.
Fig.5.C

-
WB
-
Collected and cropped from Biomedicines by CiteAb, provided under a CC-BY license
Image 1 of 41
In Cell Death Dis on 16 February 2022 by Ruan, Y., Hu, J., et al.
Fig.1.A

-
WB
-
Collected and cropped from Cell Death Dis by CiteAb, provided under a CC-BY license
Image 1 of 41
In Int J Mol Sci on 6 July 2021 by Daňhelovská, T., Zdrazilova, L., et al.
Fig.1.A

-
WB
-
Homo sapiens (Human)
Collected and cropped from Int J Mol Sci by CiteAb, provided under a CC-BY license
Image 1 of 41
In Nat Commun on 30 March 2021 by Todkar, K., Chikhi, L., et al.
Fig.5.E

-
WB
-
Collected and cropped from Nat Commun by CiteAb, provided under a CC-BY license
Image 1 of 41
In Mol Oncol on 1 December 2020 by Shi, L., Liu, J., et al.
Fig.2.A

-
WB
-
Collected and cropped from Mol Oncol by CiteAb, provided under a CC-BY license
Image 1 of 41
In J Cachexia Sarcopenia Muscle on 1 June 2020 by Shah, D. S., Nisr, R. B., et al.
Fig.7.C
-
WB
-
Collected and cropped from J Cachexia Sarcopenia Muscle by CiteAb, provided under a CC-BY license
Image 1 of 41
In J Cachexia Sarcopenia Muscle on 1 June 2020 by Shah, D. S., Nisr, R. B., et al.
Fig.4.A
-
WB
-
Collected and cropped from J Cachexia Sarcopenia Muscle by CiteAb, provided under a CC-BY license
Image 1 of 41
In Int J Mol Sci on 4 March 2020 by Duarte, A. I., Candeias, E., et al.
Fig.7.B

-
WB
-
Mus musculus (House mouse)
Collected and cropped from Int J Mol Sci by CiteAb, provided under a CC-BY license
Image 1 of 41
In Ann Clin Transl Neurol on 1 January 2020 by Verdura, E., Schlüter, A., et al.
Fig.1.F

-
WB
-
Collected and cropped from Ann Clin Transl Neurol by CiteAb, provided under a CC-BY license
Image 1 of 41
In Cell Mol Life Sci on 1 December 2019 by Nisr, R. B., Shah, D. S., et al.
Fig.8.A
-
WB
-
Collected and cropped from Cell Mol Life Sci by CiteAb, provided under a CC-BY license
Image 1 of 41
In Elife on 30 October 2019 by Berndsen, K., Lis, P., et al.
Fig.3.A

-
WB
-
Collected and cropped from Elife by CiteAb, provided under a CC-BY license
Image 1 of 41