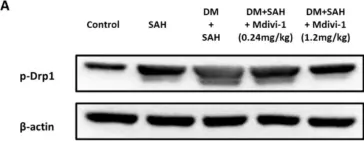
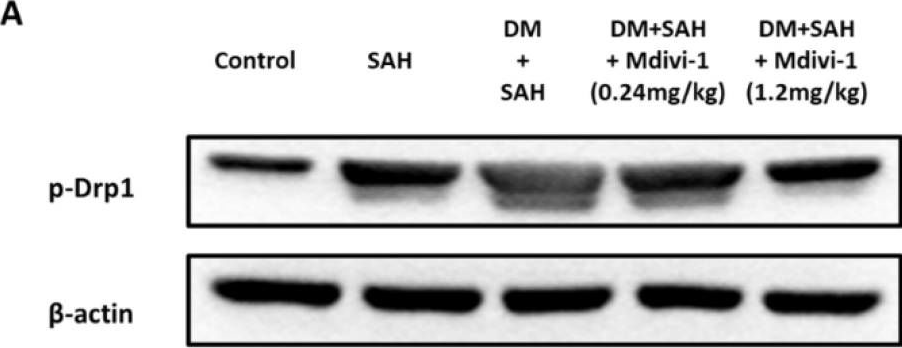
Background: Neurological deficits following subarachnoid hemorrhage (SAH) are caused by early or delayed brain injuries. Our previous studies have demonstrated that hyperglycemia induces profound neuronal apoptosis of the cerebral cortex. Morphologically, we found that hyperglycemia exacerbated late vasospasm following SAH. Thus, our previous studies strongly suggest that post-SAH hyperglycemia is not only a response to primary insult, but also an aggravating factor for brain injuries. In addition, mitochondrial fusion and fission are vital to maintaining cellular functions. Current evidence also shows that the suppression of mitochondrial fission alleviates brain injuries after experimental SAH. Hence, this study aimed to determine the effects of mitochondrial dynamic modulation in hyperglycemia-related worse SAH neurological prognosis. Materials and methods: In vitro, we employed an enzyme-linked immunosorbent assay (ELISA) to detect the effect of mitochondrial division inhibitor-1 (Mdivi-1) on lipopolysaccharide (LPS)-induced BV-2 cells releasing inflammatory factors. In vivo, we produced hyperglycemic rats via intraperitoneal streptozotocin (STZ) injections. Hyperglycemia was confirmed using blood-glucose measurements (>300 mg/dL) 7 days after the STZ injection. The rodent model of SAH, in which fresh blood was instilled into the craniocervical junction, was used 7 days after STZ administration. We investigated the mechanism and effect of Mdivi-1, a selective inhibitor of dynamin-related protein (Drp1) to downregulate mitochondrial fission, on SAH-induced apoptosis in a hyperglycemic state, and evaluated the results in a dose−response manner. The rats were divided into the following five groups: (1) control, (2) SAH only, (3) Diabetes mellitus (DM) + SAH, (4) Mdivi-1 (0.24 mg/kg) + DM + SAH, and (5) Mdivi-1 (1.2 mg/kg) + DM + SAH. Results: In vitro, ELISA revealed that Mdivi-1 inhibited microglia from releasing inflammatory factors, such as tumor necrosis factor-α (TNF-α), interleukin (IL)-1β, and IL-6. In vivo, neurological outcomes in the high-dose (1.2 mg/kg) Mdivi-1 treatment group were significantly reduced compared with the SAH and DM + SAH groups. Furthermore, immunofluorescence staining and ELISA revealed that a high dose of Mdivi-1 had attenuated inflammation and neuron cell apoptosis by inhibiting Hyperglycemia-aggravated activation, as well as microglia and astrocyte proliferation, following SAH. Conclusion: Mdivi-1, a Drp-1 inhibitor, attenuates cerebral vasospasm, poor neurological outcomes, inflammation, and neuron cell apoptosis following SAH + hyperglycemia.
Product Citations: 2
Applications
Reactivity
Research Area
In International Journal of Molecular Sciences on 22 June 2022 by Chung, C. L., Huang, Y. H., et al.
-
WB
-
Cell Biology
In Journal of the American Heart Association: Cardiovascular and Cerebrovascular Disease on 16 October 2018 by Abdullah, C. S., Alam, S., et al.
Background The Sigma 1 receptor (Sigmar1) functions as an interorganelle signaling molecule and elicits cytoprotective functions. The presence of Sigmar1 in the heart was first reported on the basis of a ligand-binding assay, and all studies to date have been limited to pharmacological approaches using less-selective ligands for Sigmar1. However, the physiological function of cardiac Sigmar1 remains unknown. We investigated the physiological function of Sigmar1 in regulating cardiac hemodynamics using the Sigmar1 knockout mouse (Sigmar1-/-). Methods and Results Sigmar1-/- hearts at 3 to 4 months of age showed significantly increased contractility as assessed by left ventricular catheterization with stimulation by increasing doses of a β1-adrenoceptor agonist. Noninvasive echocardiographic measurements were also used to measure cardiac function over time, and the data showed the development of cardiac contractile dysfunction in Sigmar1 -/- hearts as the animals aged. Histochemistry demonstrated significant cardiac fibrosis, collagen deposition, and increased periostin in the Sigmar1 -/- hearts compared with wild-type hearts. Ultrastructural analysis of Sigmar1-/- cardiomyocytes revealed an irregularly shaped, highly fused mitochondrial network with abnormal cristae. Mitochondrial size was larger in Sigmar1-/- hearts, resulting in decreased numbers of mitochondria per microscopic field. In addition, Sigmar1-/- hearts showed altered expression of mitochondrial dynamics regulatory proteins. Real-time oxygen consumption rates in isolated mitochondria showed reduced respiratory function in Sigmar1-/- hearts compared with wild-type hearts. Conclusions We demonstrate a potential function of Sigmar1 in regulating normal mitochondrial organization and size in the heart. Sigmar1 loss of function led to mitochondrial dysfunction, abnormal mitochondrial architecture, and adverse cardiac remodeling, culminating in cardiac contractile dysfunction.
-
WB
-
Mus musculus (House mouse)
-
Cardiovascular biology
-
Cell Biology
In Int J Mol Sci on 22 June 2022 by Chung, C. L., Huang, Y. H., et al.
Fig.7.A
-
WB
-
Collected and cropped from International Journal of Molecular Sciences by CiteAb, provided under a CC-BY license
Image 1 of 1